Introduction
Cardiomyopathies are a group of severe diseases. On most occasions, they represent genetical damages of the myocardium, in which its structure and function are impaired; hence, heart failure syndrome develops [1]. In 2015, cardiomyopathy and myocarditis affected 2.5 million people [2].
CA is found in 2.3% of deceased under the age of 50 years old; however, in 50-70 years old people, its prevalence is 30%; in the age group of 70-80 years old, it occurs in 41% of patients; and in those who died over the age of 90, amyloidosis was found in 71-90% of cases. Aortic amyloidosis is less common, but its incidence also increases with age, reaching 53% after 90 years of age. Severe damage and disability, lack of effective preventive measures and treatment, and diversity of visceral manifestations, as well as insufficient knowledge of its etiology and pathogenesis, determine the relevance of studying amyloidosis in general, and its cardiopathic type in particular. With the development of microcirculatory disorders, amyloid can be deposited in any cardiac structure, including pericardium, valves, myocardial stroma, endocardium, papillary muscles, and small vessels of the myocardium. However, at autopsy, it could be diagnosed only by microscopy rather than macroscopically [3].
Transthyretin (TTR, thyroxine-binding prealbumin with molecular weight of 55 kDa) is a profuse protein produced by the liver. It functions as a transporter of thyroxine and retinol. It circulates predominantly as a homotetramer, with a small amount of TTR circulating in monomeric form. The monomeric form of TTR is prone to abnormal folding and is gradually deposited as amyloid deposits.
Until a few years ago, TTR was used in clinical practice as a unique marker to identify several pathological conditions. Thus, TTR serum concentration could be used as a differential characteristic of a variety of nutritional/inflammatory processes. Moreover, as an etiological marker, TTR belongs to the group of human-derived proteins related to amyloidosis via the formation and aggregation of improperly folded proteins [4].
The abnormal folding of TTR, including limiting tetramer dissociation and partial monomer denaturation, is sufficient for TTR misassembly into amyloid and other abnormal quaternary structures, associated with senile systemic amyloidosis, familial amyloid polyneuropathy, and familial amyloid cardiomyopathy.
Two main subtypes of TTR amyloidosis (ATTR) exist: wild-type ATTR and mutant ATTR. In the former, TTR is a normal (nonmutated) protein, and over a period of decades, it gradually sediments in the form of amyloid deposits. Although small amounts of deposits may occur in the soft tissue (causing carpal tunnel syndrome) and vasculature, the primary pathologic deposits occur in the heart.
Patients with mutant ATTR are born with a pathologic mutation in the TTR gene, leading to accelerated amyloid deposition. Most commonly, mutant ATTR is deposited in the heart and/or nerves, with the pattern of deposition largely dependent on the mutation. In the United States, the most commonly encountered mutation is by far V122I mutation: it is present in 3-3.5% of individuals of African descent. The exact penetrance of the mutation is unknown (most likely, it is <10% [5]), but it is almost certainly a severely underdiagnosed form of heart failure in the African American population.
Prognosis in amyloidosis is dependent primarily on the degree of cardiac involvement. In amyloid light-chain amyloidosis (AL amyloidosis, also known as primary amyloidosis, which is the most widespread form of systemic amyloidosis), the diagnosis depends on circulating light-chain levels. Because chemotherapy options have greatly expanded in recent years, the prognosis for AL amyloidosis, is has markedly improved as well, with the life expectancy of most patients, including many of those with significant cardiac involvement, measured in years rather than months [6, 7]. The prognosis in ATTR is generally more favorable than in AL amyloidosis, even though both forms of the disease result in high annual mortality.
Treatment options for cardiac amyloidosis
Over the past decade, chemotherapy options for AL amyloidosis have markedly improved, so that most patients are able to achieve substantial reduction in their circulating pathologic light chains, or complete normalization. Chemotherapy typically involves the combination of multiple classes of antineoplastics, including alkylators (e.g., melphalan or cyclophosphamide), steroids (for instance, dexamethasone), proteasome inhibitors (for example, bortezomib or carfilzomib), and/or immunomodulators (such as lenalidomide or pomalidomide). An alternative strategy involves autologous stem cell transplant utilizing high-dose chemotherapy with an alkylating agent. Although autologous stem cell transplant can be an effective method of lowering light chains and is used by many medical centers, the only randomized trial comparing it with conventional chemotherapy has established that autologous stem cell transplant had inferior outcomes [8].
Because ATTR is not a malignant process, chemotherapy is of no use. Although there are no currently approved disease-modifying medications for ATTR, multiple agents were studied and/or are in the late-phase clinical trials. These include the following.
Diflunisal [5-(2,4-difluorophenyl) salicylic acid] (Figure 1a) is a nonsteroidal anti-inflammatory drug (NSAID) used, in general, for treating acute and chronic inflammatory diseases associated with various pains, caused by osteoarthritis, rheumatoid joint inflammation, essential dysmenorrhea, etc. [9].
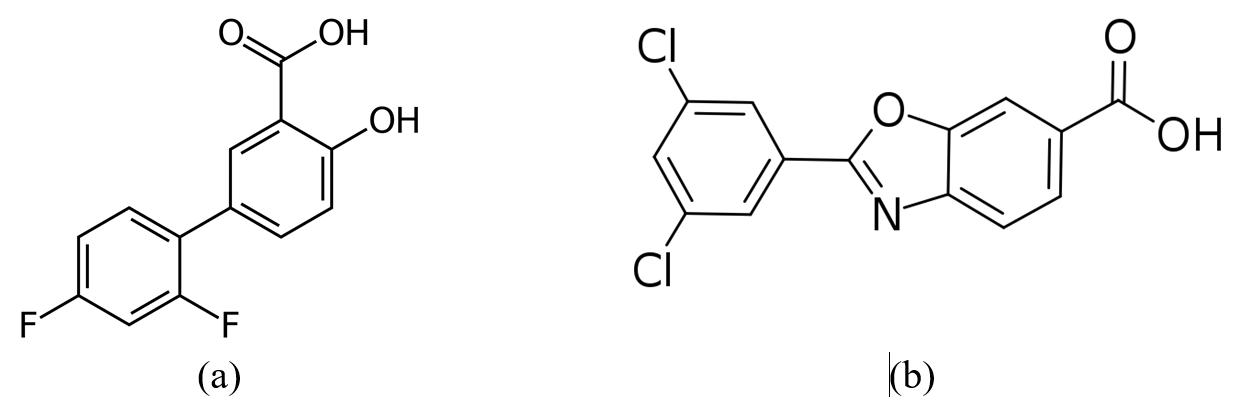
Figure 1. Structure of diflunisal (a) and tafamidis (b).
It was established that this NSAID can also stabilize the tetrameric form of TTR. A randomized trial demonstrated slowing down the disease progression among patients with polyneuropathy due to mutant ATTR [10]. Because NSAIDs are somewhat contraindicated in heart failure, this drug is not likely to be a good option for ATTR cardiomyopathy. Furthermore, in spite of prolonged therapeutic effect, the oral use of diflunisal causes serious gastrointestinal side effects, such as peptic ulcer disease, bleeding, kidney damage, digestive disorder, gastrointestinal distress, etc. [11].
Tafamidis (Figure 1b) is an oral form (Vydagel) approved in some parts of the world (Europe and Japan) for mutant ATTR, causing polyneuropathy, but it is not approved in the United States. A phase 3 study investigating its usefulness in ATTR cardiomyopathy (both wild-type ATTR and mutant ATTR) has completed an enrollment [12]. Tafamidis is an effective and safe oral medication for the treatment of cardiomyopathy of TTR-mediated amyloidosis in adults, in order to reduce their cardiovascular mortality and cardiovascular-related hospitalization rates [28].
Two agents have been investigated that work via siRNA or RNA interference (decreasing production of TTR by the liver) are in phase 3 trials for ATTR, including both polyneuropathy and cardiomyopathy forms [13,14].
In October of 2016, a phase III clinical trial of revusiran (Figure 2) was halted due to increased mortality, and further development of the compound was discontinued [15, 16].
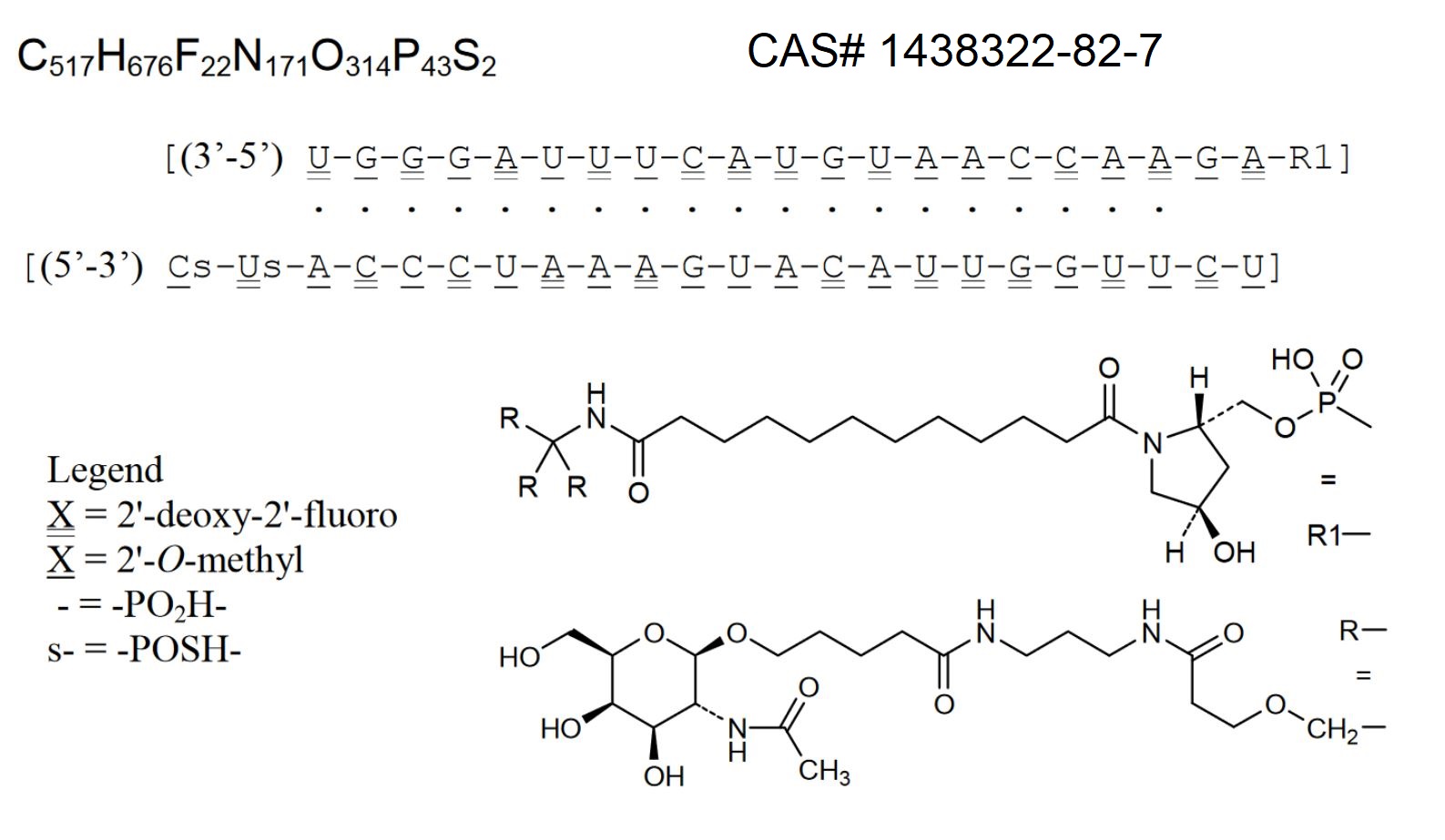
Figure 2. Structure of revusiran.
Patisiran (its trade name is Onpattro™, Figure 3) was approved for medical use to treat hereditary TTR-mediated amyloidosis in the United States and the European Union in August 2018 [17-19].
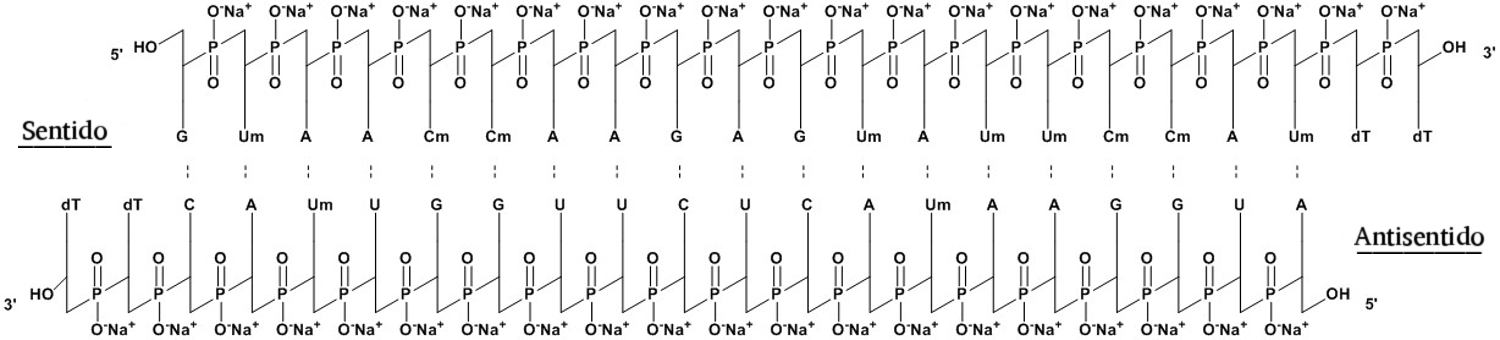

Figure 3. Structure of patisiran.
Patisiran is a liposomal siRNA. It specifically reduces the accumulation of TTR in tissues, which results in an improvement of both neuropathy and cardiac function. Patisiran is considered a prototype for smart drugs (nootropics) used in targeted therapy.
Patisiran is administered intravenously once every three weeks, significantly reducing TTR content and improving the patient’s neuropathy and quality of life. It demonstrated an average TTR knockdown of 87% and a maximum TTR protein knockdown of 96%. Although clinical trials implied its relatively high toleration, its common side effects include tract infections, infusion reactions [19, 20], as well as a reduction in the vitamin А level [21].
An important drawback of patisiran is the high cost of treatment, estimated at approximately $450,000 [22, 23].
Drug delivery systems for medications against cardiac amyloidosis
The use of a drug delivery system (DDS), which may include polymer nanoparticles, layer-by-layer capsules, dendrimers, carbon nanotubes, micelles, liposomes, is always required to enhance the efficacy and safety of therapeutic agents. It allows reducing the dose and, as a result, minimizing side effects. Nanoscale materials impact the kinetics of medicines in vivo, depending on pathophysiological/physiological mechanisms, such as vascular permeability and assimilation by the mononuclear phagocyte system, which constitute passive targeting properties of nano-DDS. On the contrary, the active targeting strategy employs a specific targeting structure for nano-DDS that binds to the target molecule specific for a certain disease process, such as tumor-specific antigens and induction of adhesion molecules [24-27].
The DDSs for tafamidis, being the first-in-its-class TTR stabilizer for TTR amyloid cardiomyopathy, are not developed as yet [28].
Revusiran is a non-genotoxic and non-carcinogenic in vitro and in vivo agent without adverse side effects in rats at higher doses, being siRNA of the first generation, targeting TTR conjugated to the N-acetylgalactosamine (GalNAc) ligand to facilitate delivery to hepatocytes via absorption by the asialoglycoprotein receptors. Revusiran, which is under development for treating hereditary TTR-mediated amyloidosis, has been discontinued due to an imbalance in mortality in the ‘ENDEAVOUR’ (phase 3) clinical trial. Nonclinical safety assessment included safety pharmacology, acute and repeat-dose toxicity, genotoxicity, and carcinogenicity. There were no effects on cardiovascular or respiratory function revealed in monkeys after single doses of up to 100 mg/kg. No neurological effects were noted in monkeys in repeat-dose studies of up to 300 mg/kg [29].
Patisiran is a siRNA encapsulated within a lipid nanoparticle delivery system for targeted delivery to the liver, which is the primary source of serum TTR [30-32].
Other compounds under development
With regard to other pharmaceutical drugs, which are being developed recently, we found a study, in which monovalent small molecules that bind to one or both of the unoccupied thyroid hormone binding sites at the border of the TTR quaternary structure stabilized the native state, thereby increasing the kinetic barrier for tetramer dissociation sufficient for the rate of dissociation; and, as a result, amyloidosis was developing slowly. Bivalent inhibitors of amyloidosis bind to both binding sites simultaneously. The candidate bivalent inhibitors are generally unable to bind to the native TTR tetramer and typically do not engage in monovalent binding because of their strong inhibitor orientation preferences. The TTR quaternary structure assembles around several bivalent inhibitors if the inhibitor intercepts the protein before the assembly. Some of the wild-type TTR bivalent inhibitor complexes retain a tetrameric structure when subjected to substantial denaturation stresses. The superior bivalent inhibitor reduced the acid-mediated formation of TTR amyloid fibrils to 6% of the formation exhibited by TTR in the absence of the inhibitor, which was a significant improvement over approximately 30% observed for superior monovalent inhibitors. The apparent dissociation rate of the best bivalent inhibitor was virtually zero, which was consistent with the idea that dissociation of the TTR tetramer and dissociation of the inhibitor were linked as a result of the assembly of the tetramer to form the inhibitor. The X-ray co-crystal structures of two complexes demonstrated that the bivalent inhibitors simultaneously occupied both sites in the TTR, which corresponded to a 1:1 binding stoichiometry obtained from high-performance liquid chromatography (HPLC) analysis. The goal of our study was to demonstrate that bivalent inhibitors could be beneficial, resulting in the best inhibitors produced to date. Accordingly, the molecules capable of intercepting TTR during folding and assembly in the lumen of the endoplasmic reticulum are of obvious interest [33].
Conclusion
The choice of medications for treating CA is extremely limited, and all of them have non-selective action modes causing substantial side effects. The search for new preparations against CA, along with the development of DDSs aimed at increasing the selectivity of action are crucial fields in cardiology.
Conflict of interest
Not declared.
Acknowledgments
This study was supported by the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075-15-2021-1349).
- d'Amati G, Giordano C. Chapter 11 – Cardiomyopathies. In: Buja LM, Butany J. Cardiovascular Pathology. 4th ed. Academic Press 2016; 435-483. https://doi.org/10.1016/B978-0-12-420219-1.00011-2.
- GBD 2015 Disease and Injury Incidence and Prevalence, Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2015; 388(10053): 1545-1602. https://doi.org/10.1016/s0140-6736(16)31678-6.
- Nuvolone M, Milani P, Palladini G, Merlini G. Management of the elderly patient with AL amyloidosis. Eur J Intern Med 2018; 58: 48-56. https://doi.org/10.1016/j.ejim.2018.05.004.
- Buxbaum JN, Reixach N, Transthyretin: The servant of many masters. Cell Mol Life Sci 2009; 66(19): 3095-3101. https://doi.org/10.1007/s00018-009-0109-0.
- Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med 2015; 372(1): 21-29. https://doi.org/10.1056/nejmoa1404852.
- Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: A staging system for primary systemic amyloidosis. J Clin Oncol 2004; 22(18): 3751-3717. https://doi.org/10.1200/jco.2004.03.029.
- Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30(9): 989-995. https://doi.org/10.1200/jco.2011.38.5724.
- Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med 2007; 357(11): 1083-1093. https://doi.org/10.1056/nejmoa070484.
- Sallam MA, Motawaa AM, Mortada SM. An insight on human skin penetration of diflunisal: Lipogel versus hydrogel microemulsion. Drug Dev Ind Pharm 2015; 41(1): 141-147. https://doi.org/10.3109/03639045.2013.850711.
- Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013; 310(24): 2658-2667. https://doi.org/10.1001/jama.2013.283815.
- Kaur A, Goindi Sh, Katare OP. Formulation, characterization and in vivo evaluation of lipid-based nanocarrier for topical delivery of diflunisal. J Microencapsulation 2016; 33(5): 475-486. https://doi.org/10.1080/02652048.2016.1216189.
- Safety and Efficacy of Tafamidis in Patients with Transthyretin Cardiomyopathy (ATTR-ACT). NCT01994889. 2019. https://clinicaltrials.gov/ct2/show/NCT01994889?term=NCT01994889&rank=1.
- ENDEAVOUR: Phase 3 Multicenter Study of Revusiran (ALN-TTRSC) in Patients with Transthyretin (TTR) Mediated Familial Amyloidotic Cardiomyopathy (FAC). NCT02319005. (National Institutes of Health website). 2018. https://clinicaltrials.gov/ct2/show/NCT02319005?term=NCT02319005&rank=1.
- Efficacy and Safety of Inotersen in Familial Amyloid Polyneuropathy. NCT01737398. 2019. https://clinicaltrials.gov/ct2/show/NCT01737398?term=NCT01737398&rank=1. APOLLO: The Study of an Investigational Drug, Patisiran (ALN-TTR02), for the Treatment of Transthyretin (TTR)-Mediated Amyloidosis. NCT01960348. 2018. https://clinicaltrials.gov/ct2/show/NCT01960348?term=NCT01960348&rank=1.
- Adams B. Alnylam plunges after deaths force it to pull Ph III blockbuster candidate revusiran. 2016 https://www.fiercebiotech.com/biotech/alnylam-plunges-after-deaths-sees-it-pull-phiii-blockbuster-candidate-revusiran.
- FierceBiotech. After 18 Deaths in phase III, alnylam halts revusiran development". Genetic Engineering and Biotechnology News. 2016. https://www.genengnews.com/news/after-18-deaths-in-phase-iii-alnylam-halts-revusiran-development/.
- Drug Approval Package: Onpattro (patisiran). U.S. Food and Drug Administration (FDA). 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922orig1s000toc.cfm.
- Onpattro EPAR. European Medicines Agency (EMA). 2021. https://www.ema.europa.eu/en/medicines/human/EPAR/onpattro.
- Urits I, Swanson D, Swett MC, Patel A, Berardino L, Amgalan A, et al. A Review of patisiran (ONPATTRO®) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther 2020; 9(2): 301-315. https://doi.org/10.1007/s40120-020-00208-1.
- Hawkins PN, Ando Y, Dispenzeri A, Gonzalez-Duarte A, Adams D, Suhr OB. Evolving landscape in the management of transthyretin amyloidosis. Ann Med 2015; 47(8): 625-638. https://doi.org/10.3109/07853890.2015.1068949.
- FDA Label of Patisiran. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210922s000lbl.pdf.
- Jarvis LM. The new drugs of 2018. Chem Eng News 2019; 97(3): 33-37. http://doi.org/10.1021/cen-09703-cover.
- Sekijima Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J Clin Pharm Ther 2014; 39(3): 225-233. https://doi.org/10.1111/jcpt.12145.
- Matoba T, Egashira K. Nanoparticle-mediated drug delivery system for cardiovascular disease. Int Heart J 2014; 55(4): 281-286. https://doi.org/10.1536/ihj.14-150.
- Matoba T, Koga JI, Nakano K, Egashira K, Tsutsui H. Nanoparticle-mediated drug delivery system for atherosclerotic cardiovascular disease. J Cardiol 2017; 70(3): 206-211. https://doi.org/10.1016/j.jjcc.2017.03.005.
- Katsuki S, Matoba T, Koga JI, Nakano K, Egashira K. Anti-inflammatory nanomedicine for cardiovascular disease. Front Cardiovasc Med 2017; 4: 87. https://doi.org/10.3389/fcvm.2017.00087.
- Fujiwara M, Matoba T, Koga JI, Okahara A, Funamoto D, Nakano K, et al. Nanoparticle incorporating toll-like receptor 4 inhibitor attenuates myocardial ischemia-reperfusion injury by inhibiting monocyte-mediated inflammation in mice. Cardiovasc Res 2019; 115(7): 1244-1255. https://doi.org/10.1093/cvr/cvz066.
- Park J, Egolum U, Parker Sh, Andrews E, Ombengi D, Ling H. Tafamidis: A first-in-class transthyretin stabilizer for transthyretin amyloid cardiomyopathy. Ann Pharmacother 2020; 54(5): 470-477. https://doi.org/10.1177/1060028019888489.
- Sutherland JE, Hettinger JL, Chan A, Gilbert J, Warner GL, Davis WP. Nonclinical safety profile of revusiran, a 1st-aeneration GalNAc-siRNA conjugate for treatment of hereditary transthyretin-mediated amyloidosis. Nucleic Acid Ther 2010; 30(1): 33-49. https://doi.org/10.1089/nat.2019.0796.
- Yang J. Patisiran for the treatment of hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol 2019; 12(2): 95-99. https://doi.org/10.1080/17512433.2019.1567326.
- Wahlich J, Desai A, Greco F, Hill K, Jones AT, Mrsny RJ, et al. Nanomedicines for the delivery of biologics. Pharmaceutics 2019; 11(5): 210. https://doi.org/10.3390/pharmaceutics11050210.
- Dong Y, Siegwart DJ, Anderson DG. Strategies, design, and chemistry in siRNA delivery systems. Adv Drug Deliv Rev 2019; 144: 133-147. https://doi.org/10.1016/j.addr.2019.05.004.
- Green NS, Palaninathan SK, Sacchettini JC, Kelly JW. Synthesis and characterization of potent bivalent amyloidosis inhibitors that bind prior to transthyretin tetramerization. J Am Chem Soc 2003; 125(44): 13404-13414. https://doi.org/10.1021/ja030294z.
Received 5 October 2021, Revised 26 November 2021, Accepted 10 December 2021
© 2021, Russian Open Medical Journal
Correspondence to Svetlana Morozkina. Address: Center for Chemical Engineering, ITMO University, 49 Kronverkskiy Prospect, bldg. A, Saint Petersburg 197101, Russia. Phone: +7-911-938-93-15; E-mail: Morozkina.Svetlana@gmail.com.