Introduction
Dendritic cells (DCs) are professional antigen-presenting cells (APCs) that effectively induce adaptive immunity against tumours and pathogens upon interactions with foreign antigens and danger signals. Therefore, DCs serve as a link between innate and adaptive immunity [1, 2]. DCs are widely localized throughout tissues, where they collect exogenous and endogenous antigens by cross-presentation. Based on phenotype and specialized functions, DCs are classified as conventional (cDCs) or plasmacytoid DCs (pDCs). cDCs are further grouped into cDC1s and cDC2s, depending on their ability to present antigen via MHC class I or class II, respectively [3]. cDC1s play a major role in the presentation and recognition of cancer cell antigens. These cells are responsible for antigen recognition and transport to appropriate endosomal compartments and the subsequent processing of antigens for cross-presentation to naïve CD8 T cells through MHC class I to initiate the immune response [4-7]. cDC1s can also present antigens through MHC class II and polarise CD4 T cells towards a Th1 phenotype by secreting IL-12 [8]. cDC2s have been reported to present antigens through MHC class II and activate the expansion of CD4 T cells [9, 10]. pDCs specialize in the production of large amounts of type I interferon (IFN) upon viral exposure [11, 12].
Mo-DCs are distinct DCs derived from Ly6Chi monocytes and have a different phenotype than conventional DCs derived from bone marrow precursors (mouse cDC1s – Ly6CloCD64loCD24+CD11blo; mouse cDC2s – Ly6CloCD64loCD24int-loCD11b+; mouse monocyte-derived DCs – Ly6ChiCD64hiCD24intCD11b+). Human mo-DCs have been identified in solid tumours (tumour-associated DCs (TADCs)); tumour ascites; and healthy tissues, such as the intestine and skin in vivo. Mo-TADC subsets have been found in tumour cell cultures as 3LL-R, T241, LLC-OVA, MMTV-PyMT, 3LL-S, MC38, and B16 [13].
Autologous DCs are widely used in immunotherapy and have become popular as a safe and reliable therapeutic approach against cancer. Multiple studies reported improved overall survival by approximately 20% in cancer patients when treated with DC vaccines [14-17]. Recently, the use of autologous ex vivo-derived mature DCs in combination with cytokine-induced killer (CIK) cells has become increasingly popular as a promising novel strategy for cancer therapy.
Typically, immune cells recognize specific antigens presented by MHC molecules on infected cell surfaces, triggering the release of cytokines that subsequently cause lysis or apoptosis. CIK cells, on the other hand, are able to track infected or malignant cells in the absence of antibodies and MHC, a feature that renders these cells capable of a rapid and unbiased immune response, primarily due to the increased proliferation of CD3+CD56+ double-positive cells. Thus, terminally differentiated CD3+CD56+ CIK cells are characterized by both MHC-restricted and MHC-unrestricted antitumour cytotoxicity against a broad range of cancer cells [18]. This characteristic is of particular importance as harmful cells that lack MHC markers cannot be recognized by other immune cells.
In a number of phase I and phase II studies, autologous and allogeneic CIK cells displayed high cytotoxic potential against different tumours with mild side effects. In many cases, CIK cell treatment led to complete remission of the tumour burden, prolonged survival, and improved quality of life, even in advanced disease stages. Currently, CIK cell treatment is restricted to clinical studies [19, 20]. However, in certain cases, CIK cell therapy alone has shown relatively low cytotoxicity due to a lack of tumour specificity [21, 22]. CIK cell function was also shown to be decreased in the presence of immunomodulatory regulatory/suppressor T (Treg) cells, which are elevated in cancer patients and are responsible for the inhibition of the antitumour immune response [23]. Clinical trials revealed that combination therapy with CIK cells and mo-DCs and chemotherapy significantly prolonged survival and enhanced immune function in patients. One- and two-year overall survival rates increased in patients with solid tumours who received combination therapy with CIK cells and mo-DCs compared with those who received chemotherapy alone (from 58.1% to 76.5% and from 11.2% to 28.6%, respectively) [24]. Moreover, the immune function of patients improved after combination therapy with CIK cells and mo-DCs, as demonstrated by the significant decrease in the number of CD8 cells and increase in interferon-gamma (IFN-γ) and IL-12 levels [25].
This literature review presents the mechanisms of the interaction of DCs and CIK cells with tumours and the proper methods for DC activation. This review was performed to investigate the pathways of tumour antigen presentation by human DCs and the crosstalk between DCs, CIK cells, and tumours to determine the most efficient means of combination therapy.
Current protocols for obtaining mo-DCs and CIK cells and cell characteristics
There are several cytokine cocktails that induce mo-DC differentiation in vitro. It has been reported that mo-DCs do not require GM-CSF signalling for in vivo differentiation. However, mo-DCs were shown to be heavily affected by the absence of Flt3L and the inability to respond to GM-CSF [26]. Phenotypic comparison with ascites-derived mo-DCs showed that when cultured with M-CSF, IL-4 and TNF-α or IL-34, IL-4, and TNF-α, human blood CD14+ monocytes undergo differentiation into CD1a+ mo-DCs and have similar expression of various surface markers (CD1b, CD11b, CD64, CD88, CD141, CD172a, CD206, CD226, FceRI, and MerTK), except for CD14, which was downregulated upon culture. Interestingly, the widely accepted culture system to induce monocyte differentiation using GM-CSF and IL-4 with or without TNF-α results in CD1a+ mo-DCs with a phenotype that is less similar to that of ascites-derived mo-DCs. Comparative transcriptomic analysis demonstrated that mo-DCs differentiated with M-CSF, IL-4, and TNF-α were highly similar to those differentiated with IL-34, IL-4, and TNF-α and clustered close to ascites mo-DCs. The transcriptome of mo-DCs differentiated with GM-CSF and IL-4 was closer to that of blood CD1c+ mo-DCs [26]. Although prominent in tumour antigen uptake, monocyte-derived TADCs lack strong T cell stimulatory capacity due to NO-mediated immunosuppression [13]. Mo-DCs generated in vitro using GM-CSF, IL-4, and TNF-α resemble naturally occurring peripheral blood DCs and, therefore, are more suitable for therapeutic applications compared to DCs generated by culturing either with M-CSF, IL-4 and TNF-α or IL-34, IL-4, and TNF-α [26].
CIK cells are a group of immune effector cells generated from peripheral lymphocytes and are activated ex vivo by exposure to IFN-γ, anti-CD3 antibodies, IL-1, and IL-2 to induce terminal differentiation and maturation. CIK cells consist of a heterogeneous population of >90% CD3+, >70% CD8+ T cells, >20% CD3+CD56+ cells (NK-T cells), and <5% CD3−CD56+ cells. CIK cells feature a mixed T- and NK cell-like phenotype and have several typical properties. These characteristics include easy generation ex vivo; potent cytotoxic activity (mediated by perforin and FasL) against various tumour cells [27]; and MHC-unrestricted cytotoxicity that is dependent on activating receptors, such as NKG2D, NKp30, and DNAM-1 [28].
Mechanism of antigen presentation by DCs
Tumour cells serve as sources of antigens for APCs by expresses mutated peptides on their surfaces. Therefore, understanding the mechanism of antigen processing and presentation by mo-DCs is a crucial step in the preparation of DC vaccines. To initiate an immune response to cancer, infected or transplanted cellular antigens must be displayed on the MHC I molecules of APCs. This process of acquiring and presenting the antigens of another cell by DCs is called “cross-priming” or “cross-presentation”. Naïve CD8 T cells constantly circulate through secondary lymphoid tissues [29]. cDC1s display tumour-associated antigens and transport them to the draining lymph node to prime and activate cytotoxic T cells. These cells subsequently migrate into the T cell compartments in the lymph nodes by their chemokine receptors, such as a C-C chemokine receptor type 7 (CCR7) [30]. In mice, cross-presentation is mainly performed by cDC1 in lymphoid organs. However, mo-DCs have the unique ability to cross-present antigens to CD8+ T cells directly in peripheral tissues [31]. In contrast, the cross-presentation of antigens in humans is similar for cDC1s, cDC2s, and pDCs. Human mo-DCs generated in vitro from monocytes and cultured with GM-CSF and IL-4 also show the ability to cross-present antigens. Tsing-Lee Tang-Huau et al. observed that human mo-DCs from peritoneal ascites and DCs generated in vitro from monocytes by culturing with M-CSF, IL-4, and TNFα cross-present exclusively using a vacuolar pathway identified by a pan-cathepsin inhibitor. However, only ascites-derived mo-DCs induce significant CD8+ T cell proliferation and expression of granzyme A, perforin, and IFN-γ [32]. Contradictory data exist on the pathway used for cross-presentation by in vitro-differentiated DCs derived from monocytes using GM-CSF and IL-4. Some studies reported the cross-presentation of soluble antigens via the vacuolar pathway [33] and others via the cytosolic pathway [34], while the cross-presentation of cell-associated antigens has been reported to be proteasome-dependent [35]. Therefore, further research is required to compare mo-DCs generated using GM-CSF and IL-4 with mo-DCs isolated from peripheral blood.
Many types of C-type lectin receptors can influence cross-presentation. Hence, antigens delivered through the C-type lectin receptor langerin on Langerhans cells [36], CLEC9A on BDCA3+ DCs [37], DCIR (CLEC4A) [38], and DC-SIGN or DEC-205 on monocyte-derived DCs or dermal DCs [39] mediate enhanced cross-presentation of human DCs. All human DC subsets and monocytes express the C-type lectin domain family 12, member A (CLEC12A) [40]. Antigen targeting to CLEC12A, mannose receptors, and CD40 enhances cross-presentation by human DCs in early endosomes, where the antigens are retained for longer periods compared to DEC-205-targeted antigens in late endosomes [41, 42]. MHC I receptors are loaded by exogenous antigens for cross‐presentation through vacuolar and cytosolic pathways [43]. In the vacuolar pathway, the cross‐presentation of antigens by APCs is TAP-independent and resistant to proteasome inhibitors (Figure 1) but sensitive to inhibitors of lysosomal proteolysis (in particular, cathepsin S inhibitors) [44, 45]. Therefore, endocytic compartments play key roles in both antigen processing and the subsequent peptide loading onto MHC class I molecules in the vacuolar pathway. In contrast, antigens internalized by the cytosolic pathway require TAP1/2 transporters and are sensitive to proteasome inhibitors, suggesting that antigens are degraded in the cytoplasm by the proteasome followed by loading of the proteasome-degraded peptides onto MHC I molecules [46]. To overcome the inefficiency of cross-presentation with soluble proteins, Schnurr et al. demonstrated that forming a vaccine using the tumour antigen NY-ESO-1 with antibodies or the immunostimulatory ISCOMATRIX (ISCOTEC AB) adjuvant (IMX) resulted in an efficient antigen delivery system that caused changes in antigen processing pathways. For direct antigen delivery to DCs, antigens could be coupled to antibodies or nanoparticles specific to DC receptors. After initial lysosomal antigen processing (inhibited by concanamycin B) and translocation into the cytosol (TAP-dependent), cross-presentation of the NY-ESO-1/antibody required proteolysis by the proteasome (inhibited by epoxomicin or lactacystin). In contrast, for NY-ESO-1/IMX, the MHC I epitope was generated in an alternative, proteasome-independent fashion [47], which indicates that antigen processing and loading may be regulated.
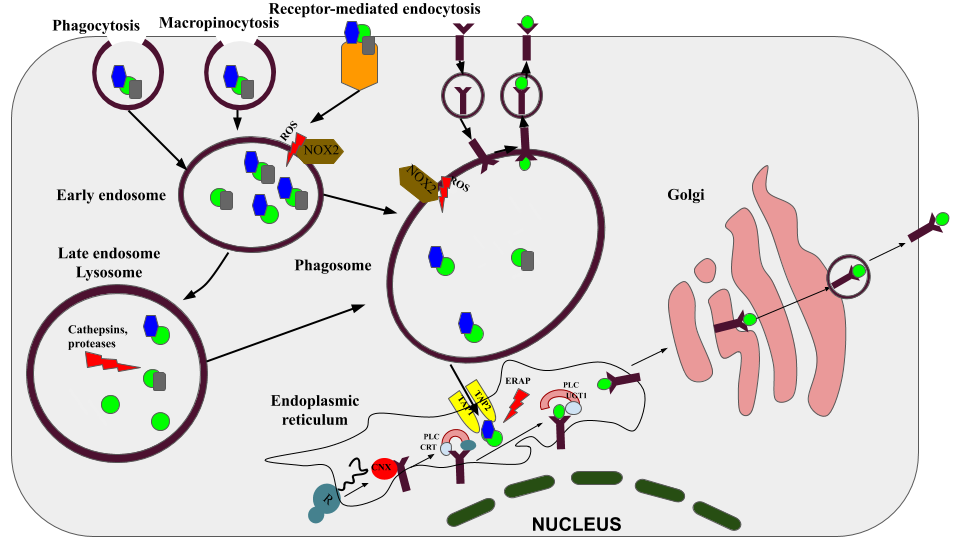
Figure 1. The molecular mechanism of cross-presentation in DCs. There are three main ways of antigens absorption occurring in DC: receptor-mediated endocytosis, phagocytosis and macropinocytosis. After properly degradation in the early endosome (by the concourse of the mild pH) or lysosome (by cathepsins and proteases), antigens can be processed in two different ways. The first is vascular, according to it, the processed antigens are directly loaded into the processed MHC-I, the whole process is carried out in the phagosome. The second pathway is the cytosolic, according to it, antigens, endogenous proteins and DRiPs are exposed by proteasomes. After processing proteins are transported by the TAP protein to the endoplasmic reticulum, where they are loaded into the MHC-I using a special peptide-loading complex. CNX, calnexin; CRT, calreticulin; UGT1, UDP-glucose: glycoprotein glucosyltransferase 1; ERAP, ER-aminopeptidases 1/2; PLC, peptide-loading complex; ERp57, protein disulfide isomerase 3; TAP1/2, transporter associated with antigen-presenting 1/2; ROS, reactive oxygen species; NOX2, NADPH oxidase 2; CLR, C-type lectins.
Therefore, antigens fused to antibodies specific to a selected DC surface receptor should mediate efficient vaccine delivery to DCs. Individual DC receptors differ widely in their expression level, internalization speed, and downstream intracellular trafficking pathways. Dec205, a molecule that is involved in late endosomal targeting, is considered a superior receptor for MHC I cross-presentation [48]. Alternatively, MHC I cross-presentation is enhanced by receptors that traffic to early, but not late, endosomes [49]. This is the case for CD40 and a mannose receptor that both traffic to early endosomes, yet for unknown reasons, CD40 is more efficient at eliciting MHC I cross-presentation [50]. A phase III clinical trial on the Muc1 fusion protein conjugated to mannan under oxidizing conditions that is recognized by the mannose receptor demonstrated a significant reduction in the recurrence rate in breast cancer patients compared to patients in the placebo group [51]. Another way to promote the APC loading of cancer antigens involves Fc receptors that interact with the Fc domains of antibodies. Hossain et al. used anti-rhamnose (Anti-Rha) antibodies to form an immune complex with a Rha-containing MUC1 vaccine in vivo for FcγR-mediated antigen uptake [52]. However, their study revealed that the antigen load, speed of internalization, surface turnover, and receptor expression level had no impact on MHC I or MHC II antigen presentation efficiency [53]. Therefore, targeting the receptor rather than the associated antigen is more likely to be the critical determinant of antigen presentation outcomes.
Maturation of DCs for DC-based vaccines
Using mature DCs is crucial for obtaining efficient DC vaccines. Foreign antigen detection and an inflammatory stimulus cause DCs to enter a complex developmental program called “maturation”. During maturation, a series of profound modifications in DC morphology and function occur. At this stage, a temporary improvement is observed in the capacity of DCs to take up antigens within 20 to 40 hours [54, 55]. This is accompanied by increased expression of co‐stimulatory molecules (CD40, CD80, and CD86) and a wide variety of inflammatory cytokines and chemokines [56]. The expression of MHC class I and class II also increases. Finally, maturation leads to the migration of DCs from tissues to the draining lymph nodes, where naïve CD8 T cell priming occurs [57].
Several practical issues involving DC maturation still have to be resolved to understand the interaction of DCs with tumour antigens. Lipopolysaccharide (LPS) in complex with cancer antigens has been widely used as a popular method to induce the maturation of DCs. The active lipid A component of LPS from Gram-negative bacteria is recognized by TLR4 in conjunction with MD2, CD14, and an LPS-binding protein [58, 59]. Pharmacological studies have reported that the recognition of PAMPs and DAMPs (but not tumours) by TLRs stimulates the production of mediators, such as type I interferon. A broad range of clinical trials have studied the adjuvant activity of TLR4 activators in vaccines based on tumour antigens. Although LPS is the most studied immunostimulatory TLR4 ligand, it is a highly toxic molecule, which hinders its use as a vaccine adjuvant. Several studies have demonstrated that the TLR4 agonist LPS induces PD-L1 on DCs and contributes to the development of tolerogenic DCs [60, 61]. Few TLR agonists have been approved by the FDA for clinical trials for TLR agonist-based immunotherapy. Bacillus Calmette-Guerin and imiquimod have been approved as standalone therapies, whereas monophosphoryl lipid A (MPL) has been approved as a vaccine component. TLR4 ligation by MPL has been shown to stimulate the tolerogenic properties of oral mucosal Langerhans cells [62]. Topical imiquimod has been shown to induce PD-L1 and CD86 in skin DC subsets [63]. TLR agonists have immune inhibitory effects, which explains the underperformance of TLR agonists as cancer therapeutics. These data suggest that the use of LPS and its components in DC maturation would be inefficient.
Another way to stimulate DC responses is to activate heat-shock proteins (HSPs). Several studies have revealed that human DCs loaded with tumour cells that were heat-treated at 42°C before being killed showed more efficient cross-priming to naïve human CD8+ T cells than DCs loaded with tumour cells that were not heated before killing [64]. HSPs function as ubiquitous chaperones that refold nascent or denatured polypeptides [65]. HSPs can also be used as adjuvants to stimulate vaccine immunogenicity. HSPs are intracellular proteins that are released into the cellular environment upon cellular injury or necrosis, but not apoptosis. HSPs can also be actively secreted into the extracellular environment by tumour cells or released from cells undergoing necrotic lysis in response to cytotoxic lymphocytes (CTLs), natural killer (NK) cells, or viral infections [66]. HSPs are regarded as DAMPs [67]. DAMPs are signals that indicate the presence of cellular damage and are alternative ligands to PRRs. HSP70 can be released from tumour cells and stimulate a potent antitumour immune response. Free extracellular HSP70 interacts with LOX-1 receptors on DCs or associates with CD94 on NK cells [68, 69]. Other surface receptors for HSPs, including scavenger receptor A (SR‐A), CD91 receptor, TLR 2, TLR4, and CD40, are involved in the endocytosis/phagocytosis [63] of HSP70-peptide complexes that are cross‐presented by DCs on MHC I [70]. HSPs are taken up into the ER through the ABC family transport system, which involves the proteins TAP1 and TAP2. TAP1 and TAP2 form a complex that transports peptides across the ER membrane and delivers them to MHC I protein complexes [71, 72]. These MHC I complexes are transported to the cytoplasm via a vesicular system and are displayed on the DC cell surface for CD8+ T cells activation [73].
Mechanism of action of CIK cells with tumour cells
The CD3+CD56+ subset of the CIK cell fraction is the main effector group that destroys malignant cells. This ability is mainly due to receptors, such as NKG2D, NKp30, and CD56. Unlike NK cells, CIK cells poorly express or do not express NKp44 or NKp46. Antibody-blocking experiments revealed that DNAM-1, NKG2D, and NKp30 are involved in the TCR-independent recognition and killing of tumour cells. However, CIK cells retain antibody-dependent cellular cytotoxicity (ADCC) ability and TCR-mediated cytotoxicity, thus exerting their “dual-functional capability” against tumour cells [18]. NKG2D is the surface cell receptor that plays an important role in the cytotoxic activity of the main effector CD3+CD56+ subset of CIK cells. NKG2D belongs to the C-type lectin-like receptor family. NKG2D is not capable of inducing a signalling cascade; therefore, the transmembrane molecule DNAX-activating protein of 10 kDa (DAP10) acts as a molecular transmitter [74]. NKG2D and its DAP10 adapter molecule form an activating receptor complex, which can signal by recruiting phosphatidylinositol-3 kinase [75]. NKG2D ligands are stress-induced proteins that are expressed mainly on the surface of cancer cells. Structurally, stress-induced proteins belong to the family of MHC class I-related ligands (MHC class I-related chain A and B [MICA, MICB]) proteins and the six unique long 16 (UL-16)-binding proteins (ULBP1-6) [76, 77]. Cytotoxic activity was shown to be mediated by this receptor in an in vitro study on the LCL 721.221 cell line not expressing MHC class-I molecules, thus excluding TCR-mediated recognition of cancer cells [78]. The decrease in cytotoxic activity is influenced by CD4+ CD25+ T cells. In an in vivo and in vitro study, it was shown that the elimination of Tregs in the initial stage of culturing CIK cells has a significant effect on the subsequent lysing ability of effector cells. The suppressive role of CD4+ CD25+ T cells is based on inhibiting the expression of the NKG2D TGF-β1 receptor on CIK cells, which is synthesized by Tregs [79]. Although early studies have shown the inability of CIK cells to mediate ADCC [80], Cappuzzello and colleagues have recently observed in vivo and in vitro that CIK cells, namely the effector fraction of CD3+CD56+ cells, are capable of donor-dependent expression of CD16 FcγRIII induced by the addition of IgG [81] monoclonal antibodies. As shown by antibodies blocking receptors, such as NKG2D NKp30, CIKs have HLA-independent cytotoxicity against the HLA I-deficient K562 cell line. However, at the same time, such antibody blocking did not affect antitumour activity of these cells against CMV-pulsed autologous T-PHA-induced blasts, which in turn demonstrates the ability of CIK cells to mediate TCR-dependent cytotoxicity [82]. Mehta and colleagues have shown that LFA-1, an adhesion molecule expressed on the surface of CIK cells, does not play a special role in the release of cytolytic granules. However, they observed that LFA-1 plays an important role in the recognition of tumour cells with surface ligands LFA-1- ICAM-1, -2, and -3, CIK cells, and in mediating cell-to-cell-mediated cytotoxicity [83]. However, for cancer cells that do not express these ligands, the cytotoxic activity of CD3+CD56+ double-positive cells remains invariably effective [18]. As shown from a CD56 knockdown experiment in CIK cells and from monoclonal antibodies against CD56 (GPR165), the CD56 receptor has significant importance in the process of recognition and lysis of target cells that express this marker. In the same experiment, it was shown that the 140-kDa isoform of CD56 is characteristic not only of CIK cells, but also of NK cells [84].
As previously mentioned, CIK cells possess properties inherent in both T cells and NK cells. Therapy based on CIK cells has many advantages. CIK cells are easily expandable in vitro; do not require exogenous administration of IL-2; and have easily manageable side effects, such as fever, headache, and rash.
Crosstalk of DCs with CIK cells
DCs are the major antigen-presenting cells and can capture and process tumour antigens and activate the immune functions of CD4+ and CD8+ T cells, NK cells, B cells, and CIK cells [85]. Subsequently, the interaction between DCs and CIK cells has been described to have a costimulatory effect on both populations, with a dramatic increase in IL-12 secretion by DCs and a significant increase in the cytotoxic activity of CIK cells [86]. Accumulating evidence indicates that coculture with in vitro-matured DCs can be used to overcome tumour-related immunosuppression and improve the antitumour efficacy of CIK cells. This effect is mediated by decreasing the number and function of CD4+CD25+ Tregs, which shows a negative correlation with IL-10 concentration and results in an enhanced expansion and frequency of CD3+CD56+ cells in the amplified cell population. However, immature DCs were reported to be responsible for the tolerance and induction of Treg cells [87]. Pasare et al. (2003) reported that DCs could block suppressor Treg activity by secreting IL-6 [88].
In many cases, an antigen-specific T cell response cannot be initiated due to the absence of functional DCs in patients with tumours [89]. Recently, DCs have been used in several studies to increase the cytotoxic activity of CIK cells in vitro or in vivo [86, 87], where tumour antigens have been coincubated with DCs in vitro.
The contact-dependent mechanism of T cell activation, where DCs contact T cells through CD80/86 and undergo cytoskeletal reorientation induced by T cells via CD40-CD40 L signalling, is well studied [90]. However, the exact mechanism of DC recognition by CIK cells is not fully understood. Lee et al. (2016) demonstrated a contact-dependent activation of CIK cells cocultured with tumour lysate–pulsed dendritic cells (tDCs) via CCR5 signalling, where tDCs express high levels of CCL5 and bind CCR5 expressed on CIK cells. Interestingly, tDCs were shown to exhibit more frequent and long-term contacts with Ccr5+/+ CIK cells than Ccr5−/− CIK cells at the single-cell level, which resulted in increased antitumour activity of Ccr5+/+ CIK cells in vitro and in vivo [91]. Another study revealed decreased cytolytic activity of CIK cells after blocking IL-12 released by DCs in a coculture system, thus demonstrating the importance of cytokine release in the activation of CIK cells. On the other hand, cellular interactions were reported to cause changes in the pattern of surface molecule expression on both DCs (increase in DC-specific, costimulatory, and antigen-presenting molecules) and CIK cells (higher levels of CD4, CD28, and CD40L surface markers), leading to an increase in IL-12 concentration and a further increase in the cytolytic capacity of the latter [86].
Conclusion
Сurrent DC-based vaccine therapy suggests different methods of DC maturation. Here, we discussed several aspects of DC maturation. Several theoretical aspects are very important. For example, linking antigens to antibodies specific to DC receptors or adjuvants (such as IMX) may stimulate different antigen processing by DCs. Induction of DC maturation with LPS or HSPs may induce the recruitment of myeloid suppressor cells and regulatory T cells by tolerogenic DCs or enhance the vaccine by additional pathways. This review revealed the role of both contact-dependent activation of CIK cells (involving CCR5 signalling) and the cytokine-dependent cytolytic capacity of CIK cells upon coculture with mature DCs. Therefore, the possibility of overcoming tumour-related immunosuppression due to the enhanced cytotoxicity and proliferation of CIK cells after coculture with in vitro-modified DCs makes such combination therapy an attractive immunomodulatory approach for the induction of anticancer immune responses.
Acknowledgment
Supported by a grant AP05135467 "Development of production technology of dendritic vaccines and cytokine-induced killer cells for combination therapy in colorectal cancer" from the Ministry of Education and Science of the Republic of Kazakhstan.
The authors would like to thank Alexander Shustov, Candidate of Biological Sciences and Head of the Laboratory of Genetic Engineering at the National Center for Biotechnology, for stimulating the idea and advice. Ruslan Kalendar, Candidate of Biological Sciences, adjunct professor at University of Helsinki, and Head of the Laboratory of Plant Genomics and Bioinformatics at the National Center for Biotechnology, is particularly acknowledged for his dedicated help in proofreading and professional editing of the paper.
Conflicts of interest
The authors declare that they have no conflicts of interest.
- Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol 2004; 5(10): 971-974. https://doi.org/10.1038/ni1004-971.
- Worbs T, Hammerschmidt SI, Forster R. Dendritic cell migration in health and disease. Nat Rev Immunol 2017; 17(1): 30-48. https://doi.org/10.1038/nri.2016.116.
- Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 2013; 31: 563-604. https://doi.org/10.1146/annurev-immunol-020711-074950.
- Cruz FM, Colbert JD, Merino E, Kriegsman BA, Rock KL. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu Rev Immunol 2017; 35: 149-176. https://doi.org/10.1146/annurev-immunol-041015-055254.
- Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. Tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 2012; 37(1): 60-73. https://doi.org/10.1016/j.immuni.2012.04.012.
- Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+cells as homologues of mouse CD8+dendritic cells. J Exp Med 2010; 207(6): 1273-1281. https://doi.org/10.1084/jem.20100348.
- Blander JM. Regulation of the cell biology of antigen cross-presentation. Annu Rev Immunol 2018; 36: 717-753. https://doi.org/10.1146/annurev-immunol-041015-055523.
- Soares H, Waechter H, Glaichenhaus N, Mougneau E, Yagita H, Mizenina O, et al. A subset of dendritic cells induces CD4+T cells to produce IFN-γ by an IL-12–independent but CD70-dependent mechanism in vivo. J Exp Med 2007; 204(5): 1095-1106. https://doi.org/10.1084/jem.20070176.
- Anderson DA, Murphy KM, Briseño CG. Development, diversity, and function of dendritic cells in mouse and human. Cold Spring Harb Perspect Biol 2017; a028613. https://doi.org/10.1101/cshperspect.a028613.
- Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology 2018; 154(1): 3-20 https://doi.org/10.1111/imm.12888.
- Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol 2004; 5(12): 1219-1226. https://doi.org/10.1038/ni1141.
- Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999; 284(5421): 1835-1837. https://doi.org/10.1126/science.284.5421.1835.
- Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun 2016; 7: 13720. https://doi.org/10.1038/ncomms13720.
- Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol 2003; 15(2): 138-147. https://doi.org/10.1016/s0952-7915(03)00015-3
- Banchereau J, Paczesny S, Blanco P, Bennett L, Pascual V, Fay J, et al. Dendritic cells: controllers of the immune system and a new promise for immunotherapy. Ann N Y Acad Sci 2003; 987(1): 180-187. https://doi.org/10.1111/j.1749-6632.2003.tb06047.x.
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12(4): 265-277. https://doi.org/10.1038/nrc3258.
- Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human dendritic cells induce the differentiation of Interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity 2009; 31(1): 158-169. https://doi.org/10.1016/j.immuni.2009.04.016.
- Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, et al. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood 2011; 118(12): 3301-3310. https://doi.org/10.1182/blood-2011-02-336321.
- Valgardsdottir R, Capitanio C, Texido G, Pende D, Cantoni C, Pesenti E, et al. Direct involvement of CD56 in cytokine-induced killer–mediated lysis of CD56+ hematopoietic target cells. Exp Hematol 2014; 42(12): 1013-1021.e1. (https://doi.org/10.1016/j.exphem.2014.08.005.
- Schmeel LC, Schmeel FC, Coch C, Schmidt-Wolf IG. Cytokine-induced killer (CIK) cells in cancer immunotherapy: report of the international registry on CIK cells (IRCC). J Cancer Res Clin Oncol 2014; 141(5): 839-849. https://doi.org/10.1007/s00432-014-1864-3.
- Hontscha C, Borck Y, Zhou H, Messmer D, Schmidt-Wolf IG. Clinical trials on CIK cells: first report of the international registry on CIK cells (IRCC). J Cancer Res Clin Oncol 2010; 137(2): 305-310. https://doi.org/10.1007/s00432-010-0887-7.
- Qiu Y, Yun MM, Dong X, Xu M, Zhao R, Han X, et al. Combination of cytokine-induced killer and dendritic cells pulsed with antigenic α-1,3-galactosyl epitope–enhanced lymphoma cell membrane for effective B-cell lymphoma immunotherapy. Cytotherapy 2016; 18(1): 91-98. https://doi.org/10.1016/j.jcyt.20.15.09.012
- Zou Y, Li F, Hou W, Sampath P, Zhang Y, Thorne SH. Manipulating the expression of chemokine receptors enhances delivery and activity of cytokine-induced killer cells. Br J Cancer 2014; 110(8): 1992-1999. https://doi.org/10.1038/bjc.2014.140.
- Zhou X, Mo X, Qiu J, Zhao J, Wang S, Zhou C, et al. Chemotherapy combined with dendritic cell vaccine and cytokine-induced killer cells in the treatment of colorectal carcinoma: a meta-analysis. Cancer Manag Res 2018; 10: 5363-5372. https://doi.org/10.2147/cmar.s173201.
- Mu Y, Zhou CH, Chen SF, Ding J, Zhang YX, Yang YP, et al. Effectiveness and safety of chemotherapy combined with cytokine-induced killer cell /dendritic cell-cytokine-induced killer cell therapy for treatment of gastric cancer in China: A systematic review and meta-analysis. Cytotherapy 2016; 18(9): 1162-1177. https://doi.org/10.1016/j.jcyt.2016.05.015.
- Goudot C, Coillard A, Villani A, Gueguen P, Cros A, Sarkizova S, et al. Aryl hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages. Immunity 2017; 47(3): 582-596.e6. https://doi.org/10.1016/j.immuni.2017.08.016.
- Schmidt TL, Negrin RS, Contag CH. A killer choice for cancer immunotherapy. Immunol Res 2014; 58(2-3): 300-306. https://doi.org/10.1007/s12026-014-8507-2.
- Pievani A, Belussi C, Klein C, Rambaldi A, Golay J, Introna M. Enhanced killing of human B-cell lymphoma targets by combined use of cytokine-induced killer cell (CIK) cultures and anti-CD20 antibodies. Blood 2010; 117(2): 510-518. https://doi.org/10.1182/blood-2010-06-290858.
- Mora JR, von Andrian UH. T-cell homing specificity and plasticity: new concepts and future challenges. Trends Immunol 2006; 27(5): 235-243. https://doi.org/10.1016/j.it.2006.03.007.
- Johnson LA, Jackson DG. Control of dendritic cell trafficking in lymphatics by chemokines. Angiogenesis 2014; 17(2): 335-345. https://doi.org/10.1007/s10456-013-9407-0.
- Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 2008; 319(5860): 198-202. https://doi.org/10.1126/science.1151869.
- Tang-Huau TL, Gueguen P, Goudot C, Durand M, Bohec M, Baulande S, et al. Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat Commun 2018; 9(1): 2570. https://doi.org/10.1038/s41467-018-04985-0.
- Ma W, Zhang Y, Vigneron N, Stroobant V, Thielemans K, van der Bruggen P, et al. Long-peptide cross-presentation by human dendritic cells occurs in vacuoles by peptide exchange on nascent MHC class I molecules. J Immunol 2016; 196(4): 1711-1720. https://doi.org/10.4049/jimmunol.1501574.
- Baleeiro RB, Walden P. Immature human DCs efficiently translocate endocytosed antigens into the cytosol for proteasomal processing. Mol. Immunol 2017; 88: 148-154. https://doi.org/10.1016/j.molimm.2017.06.028.
- Fonteneau JF, Kavanagh DG, Lirvall M, Sanders C, Cover TL, Bhardwaj N, et al. Characterization of the MHC class I cross-presentation pathway for cell-associated antigens by human dendritic cells. Blood 2003; 102(13): 4448-4455. https://doi.org/10.1182/blood-2003-06-1801.
- Fehres CM, Kalay H, Bruijns SC, Musaafir SA, Ambrosini M, Van Bloois L, et al. Cross-presentation through langerin and DC-SIGN targeting requires different formulations of glycan-modified antigens. J Control Release 2015; 203: 67-76. https://doi.org/10.1016/j.jconrel.2015.01.040.
- Schreibelt G, Klinkenberg LJ, Cruz LJ, Tacken PJ, Tel J, Kreutz M, et al. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood 2012; 119(10): 2284-2292. https://doi.org/10.1182/blood-2011-08-373944.
- Klechevsky E, Flamar A, Cao Y, Blanck J, Liu M, O'Bar A, et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood 2010; 116(10): 1685-1697. https://doi.org/10.1182/blood-2010-01-264960.
- Saluja SS, Hanlon DJ, Sharp FA, Hong E, Khalil D, Robinson E, et al. Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen. Int J Nanomedicine 2014; 9: 5231-5246. https://doi.org/10.2147/ijn.s66639.
- Lahoud MH, Proietto AI, Ahmet F, Kitsoulis S, Eidsmo L, Wu L, et al. The C-type lectin Clec12A present on mouse and human dendritic cells can serve as a target for antigen delivery and enhancement of antibody responses. J Immunol 2009; 182(12): 7587-7594. https://doi.org/10.4049/jimmunol.0900464.
- Hutten TJ, Thordardottir S, Fredrix H, Janssen L, Woestenenk R, Tel J, et al. CLEC12A-mediated antigen uptake and cross-presentation by human dendritic cell subsets efficiently boost tumor-reactive T cell responses. J Immunol 2016; 197(7): 2715-2725. https://doi.org/10.4049/jimmunol.1600011.
- Chatterjee B, Smed-Sorensen A, Cohn L, Chalouni C, Vandlen R, Lee B, et al. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 2012; 120(10): 2011-2020. https://doi.org/10.1182/blood-2012-01-402370.
- Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol 2012; 12(8): 557-569. https://doi.org/10.1038/nri3254.
- Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 2004; 21(2): 155-165. https://doi.org/10.1016/j.immuni.2004.07.004.
- Bertholet S, Goldszmid R, Morrot A, Debrabant A, Afrin F, Collazo-Custodio C, et al. Leishmania antigens are presented to CD8+ T Cells by a transporter associated with antigen processing-independent pathway in vitro and in vivo. J Immunol 2006; 177(6): 3525-3533. https://doi.org/10.4049/jimmunol.177.6.3525.
- Kovacsovics-Bankowski M, Rock K. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 1995; 267(5195): 243-246. https://doi.org/10.1126/science.7809629.
- Schnurr M, Chen Q, Shin A, Chen W, Toy T, Jenderek C, et al. Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood 2005; 105(6): 2465-2472. https://doi.org/10.1182/blood-2004-08-3105.
- Cohn L, Chatterjee B, Esselborn F, Smed-Sörensen A, Nakamura N, Chalouni C, et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J Exp Med 2013; 210(5): 1049-1063. https://doi.org/10.1084/jem.20121251.
- Tacken PJ, Ginter W, Berod L, Cruz LJ, Joosten B, Sparwasser T, et al. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood 2011; 118(15): 4111-4119. https://doi.org/10.1182/blood-2011-04-346957.
- Chatterjee B, Smed-Sörensen A, Cohn L, Chalouni C, Vandlen R, Lee BC, et al. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 2012; 120: 2011-2020. https://doi.org/10.1182/blood-2012-01-402370.
- Vassilaros S, Tsibanis A, Tsikkinis A, Pietersz GA, McKenzie IF, Apostolopoulos V. Up to 15-year clinical follow-up of a pilot Phase III immunotherapy study in stage II breast cancer patients using oxidized mannan-MUC1. Immunotherapy 2013; 5(11): 1177-1182. https://doi.org/10.2217/imt.13.126.
- Hossain MK, Vartak A, Karmakar P, Sucheck SJ, Wall KA. Augmenting vaccine immunogenicity through the use of natural human anti-rhamnose antibodies. ACS Chem Biol 2018; 13(8): 2130-2142. https://doi.org/10.1021/acschembio.8b00312
- Reuter A, Panozza SE, Macri C, Dumont C, Li J, Liu H, et al. Criteria for dendritic cell receptor selection for efficient antibody-targeted vaccination. J Immunol 2015; 194(6): 2696-2705. https://doi.org/10.4049/jimmunol.1402535.
- Gil-Torregrosa B, Lennon-Duménil A, Kessler B, Guermonprez P, Ploegh H, Fruci D, et al. Control of cross-presentation during dendritic cell maturation. Eur J Immunol 2004; 34(2): 398-407. https://doi.org/10.1002/eji.200324508.
- West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, et al. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science 2004; 305(5687): 1153-1157. https://doi.org/10.1126/science.1099153.
- Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol 2006; 6(6): 476-483. https://doi.org/10.1038/nri1845.
- Vargas P, Maiuri P, Bretou M, Sáez PJ, Pierobon P, Maurin M, et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat Cell Biol 2015; 18(1): 43-53. https://doi.org/10.1038/ncb3284.
- Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, et al. MD-2, a Molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4. J Exp Med 1999; 189(11): 1777-1782. https://doi.org/10.1084/jem.189.11.1777.
- Kirkland TN, Finley F, Leturcq D, Moriarty A, Lee JD, Ulevitch RJ, et al. Analysis of lipopolysaccharide binding by CD14. J Biol Chem 1993; 268(33): 24818-24823. https://pubmed.ncbi.nlm.nih.gov/7693705.
- Wölfle SJ, Strebovsky J, Bartz H, Sähr A, Arnold C, Kaiser C, et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol 2011; 41(2): 413-424. https://doi.org/10.1002/eji.201040979.
- Rodríguez-García M, Porichis F, de Jong OG, Levi K, Diefenbach TJ, Lifson JD, et al. Expression of PD-L1 and PD-L2 on human macrophages is up-regulated by HIV-1 and differentially modulated by IL-10. J Leukoc Biol 2010; 89(4): 507-515. https://doi.org/10.1189/jlb.0610327.
- Allam J, Peng W, Appel T, Wenghoefer M, Niederhagen B, Bieber T, et al. Toll-like receptor 4 ligation enforces tolerogenic properties of oral mucosal Langerhans cells. J Allergy Clin Immunol 2008; 121(2): 368-374.e1. https://doi.org/10.1016/j.jaci.2007.09.045.
- Hackstein H, Hagel N, Knoche A, Kranz S, Lohmeyer J, von Wulffen W, et al. Skin TLR7 Triggering Promotes Accumulation of Respiratory Dendritic Cells and Natural Killer Cells. PLoS One 2012; 7(8): e43320. https://doi.org/10.1371/journal.pone.0043320.
- Shi H, Cao T, Connolly JE, Monnet L, Bennett L, Chapel S, et al. Hyperthermia enhances CTL cross-priming. J Immunol 2006; 176(4): 2134-2141. https://doi.org/10.4049/jimmunol.176.4.2134.
- Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol 2010; 2(12): a004390-a004390. https://doi.org/10.1101/cshperspect.a004390.
- Mambula SS, Calderwood SK. Heat Shock Protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol 2006; 177(11): 7849-7857. https://doi.org/10.4049/jimmunol.177.11.7849.
- Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-κB pathway. Int Immunol 2000; 12(11): 1539-1546. https://doi.org/10.1093/intimm/12.11.1539.
- Thériault JR, Mambula SS, Sawamura T, Stevenson MA, Calderwood SK. Extracellular HSP70 binding to surface receptors present on antigen presenting cells and endothelial/epithelial cells. FEBS Letters 2005; 579(9): 1951-1960. https://doi.org/10.1016/j.febslet.2005.02.046.
- Chen X, Tao Q, Yu H, Zhang L, Cao X. Tumor cell membrane-bound heat shock protein 70 elicits antitumor immunity. Immunol Lett 2002; 84(2): 81-87. https://doi.org/10.1016/s0165-2478(02)00042-1.
- Singh-Jasuja H, Toes RE, Spee P, Münz C, Hilf N, Schoenberger SP, et al. Cross-rresentation of Glycoprotein 96–associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J Exp Med 1999; 191(11): 1965-1974. https://doi.org/10.1084/jem.191.11.1965.
- Shepherd JC, Schumacher TN, Ashton-Rickardt PG, Imaeda S, Ploegh HL, Janeway CA, et al. TAP1-dependent peptide translocation in vitro is ATP dependent and peptide selective. Cell 1993; 74(3): 577-584. https://doi.org/10.1016/0092-8674(93)80058-m.
- Schumacher TN, Kantesaria DV, Heemels MT, Ashton-Rickardt PG, Shepherd JC, Fruh K, et al. Peptide length and sequence specificity of the mouse TAP1/TAP2 translocator. J Exp Med 1994; 179(2): 533-540. https://doi.org/10.1084/jem.179.2.533.
- Neefjes J, Momburg F, Hammerling G. Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science 1993; 261(5122): 769-771. https://doi.org/10.1126/science.8342042.
- Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell–mediated killing via a Syk-independent regulatory pathway. Nat Immunol 2003; 4(6): 557-564. https://doi.org/10.1038/ni929.
- Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999; 285(5428): 730-732. https://doi.org/10.1126/science.285.5428.730.
- Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics 2001; 53(4): 279-287. https://doi.org/10.1007/s002510100325.
- Sutherland CL, Chalupny NJ, Cosman D. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol Rev 2001; 181(1): 185-192. https://doi.org/10.1034/j.1600-065x.2001.1810115.x.
- Verneris MR, Karami M, Baker J, Jayaswal A, Negrin RS. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood 2004; 103(8): 3065-3072. https://doi.org/10.1182/blood-2003-06-2125.
- Liu A, Wang Q, Yang X, Shen S, Xu Y. Cord blood CD4+/CD25+ T cells down-regulates NKG2D on cytokine-induced killer cells. Front Biosci (Landmark Ed) 2019; 24(7): 1259-1270. https://pubmed.ncbi.nlm.nih.gov/31136978.
- Wang FS, Liu MX, Zhang B, Shi M, Lei ZY, Sun WB, et al. Antitumor activities of human autologous cytokine-induced killer (CIK) cells against hepatocellular carcinoma cells in vitro and in vivo. World J Gastroenterol 2002; 8(3): 464-468. https://doi.org/10.3748/wjg.v8.i3.464.
- Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell–mediated killing via a Syk-independent regulatory pathway. Nat Immunol 2003; 4(6): 557-564. https://doi.org/10.1038/ni929.
- Kim N, Nam Y, Im K, Lim J, Jeon Y, Song Y, et al. Robust rroduction of cytomegalovirus pp65-specific T cells using a fully automated IFN-γ cytokine capture system. Transfus Med Hemother 2017; 45(1): 13-22. https://doi.org/10.1159/000479238.
- Mehta BA, Schmidt-Wolf IG, Weissman IL, Negrin RS. Two pathways of exocytosis of cytoplasmic granule contents and target cell killing by cytokine-induced CD3+ CD56+ killer cells. Blood 1995; 86(9): 3493-3499. https://pubmed.ncbi.nlm.nih.gov/7579455.
- Li H, Yu JP, Cao S, Wei F, Zhang P, An XM, et al. CD4 +CD25 + regulatory T cells decreased the antitumor activity of cytokine-induced killer (CIK) cells of lung cancer patients. J Clin Immunol 2007; 27(3): 317-326. https://doi.org/10.1007/s10875-007-9076-0.
- Wei XC, Yang DD, Han XR, Zhao YA, Li YC, Zhang LJ, et al. Bioactivity of umbilical cord blood dendritic cells and anti-leukemia effect. Int J Clin Exp Med 2015; 8(10): 19725-19730. https://pubmed.ncbi.nlm.nih.gov/26770637.
- Märten A, Ziske C, Schöttker B, Renoth S, Weineck S, Buttgereit P, et al. Interactions between dendritic cells and cytokine-induced killer cells lead to an activation of both populations. J Immunother 2001; 24(6): 502-510. https://doi.org/10.1097/00002371-200111000-00007.
- Schmidt J, Eisold S, Büchler M, Märten A. Dendritic cells reduce number and function of CD4+CD25+ cells in cytokine-induced killer cells derived from patients with pancreatic carcinoma. Cancer Immunol, Immunother 2004; 53(11): 1018-1026. https://doi.org/10.1007/s00262-004-0554-4.
- Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003; 299(5609): 1033-1036. https://doi.org/10.1126/science.1078231.
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998; 392(6673): 245-252. https://doi.org/10.1038/32588.
- Rothoeft T, Balkow S, Krummen M, Beissert S, Varga G, Loser K, et al. Structure and duration of contact between dendritic cells and T cells are controlled by T cell activation state. Eur J Immunol 2006; 36(12): 3105-3117. https://doi.org/10.1002/eji.200636145.
- Lee HK, Kim YG, Kim JS, Park EJ, Kim B, Park KH, et al. Cytokine-induced killer cells interact with tumor lysate-pulsed dendritic cells via CCR5 signaling. Cancer Lett 2016; 378(2): 142-149. https://doi.org/10.1016/j.canlet.2016.05.020.
Received 23 April 2020, Revised 16 October 2020, Accepted 4 November 2020
© 2020, Issabekova A., Zhumabekova M., Zhunussova M., Ogay V.
© 2020, Russian Open Medical Journal
Correspondence to Assel Issabekova. Phone: +7(7172)70-75-21. E-mail: issabekova@biocenter.kz.