Introduction
Over the last two decades, it has become obvious that endothelium has organ-specific and structurally functional properties, including the regulation of perfusion, vascular tone, coagulation, inflammatory reactions, vasculogenesis, and angiogenesis [1]. Endothelial dysfunction (ED), accompanying a number of ailments, especially during their treatment, is a serious complication, associated with increased risk of mortality [2]. Although ED is an early stage of changes in the cardiovascular system, it is likely to cause further complications and the onset of specific diseases. ED has been demonstrated among the earliest complications of chemotherapy, potentially triggering cardiovascular disease even in the patients with a low initial risk. It remains a significant reason of death, despite continuous optimization of treatment protocols [1]. In addition, the pathogenetic mechanisms of this complication remain largely unexplored. Scientists suggest that increased cellular decay during chemotherapy is likely to cause systemic inflammation in patients, leading to ED development [3]. Chemotherapy with various medications launches various risk factors in the patients, such as hypertension, obesity, metabolic syndrome and dyslipidemia, all of which aggravate the vascular reserve and lead to cardiotoxic effects with a negative impact on therapy effectiveness and patient adherence [4-6].
At the same time, the attention of researchers all over the world is attracted by the mechanism of pathological angiogenesis in lymphoproliferative diseases, which is characterized by persistent proliferation of endothelial cells. In cancer biology, angiogenesis is vital for tumor growth and its subsequent metastatic potential. Recently, the idea of tumor vascularity has been investigated in the studies quantifying the density of microvessels in various nosological forms, especially, in patients with lymphoproliferative disorders [7].
Chronic lymphoproliferative diseases – a group of nosologies that include pathology caused by malignant transformation of mature lymphocytes, which leads to infiltration of lymph nodes and/or extranodal organs and is accompanied by a highly variable clinical course and prognosis. This group includes Hodgkin lymphoma and non-Hodgkin lymphomas, chronic lymphocytic leukemia (CLL), hairy cell leukemia, multiple myeloma and Waldenstrom macroglobulinemia [8]. Besides chronic lymphoproliferative disorders, acute lymphoproliferative diseases are known, in particular, acute lymphoblastic leukemia, which is the most common hemoblastosis in children. The mechanisms facilitating distribution of these nosologies are still under investigation [9], and their prevalence is quite high [10]. Existing treatment protocols allow achieving remission, but patients are not immune to relapse of the disease, and subsequent treatment courses often have greater toxicity, especially, in relation to cardiovascular system, and lower efficiency [11]. These facts imply that recurrent or refractory nature of these lymphoproliferative nosologies puts patients at risk of ED development. Thus, proaggregatory and/or proinflammatory activation of endothelium, against the background of chemotherapeutic treatment, in the long run, could lead to earlier emergence of cardiovascular pathologies. Therefore, observation of the patients from this group is crucial.
The objective of this review is to present the problems and prospects of revealing early subclinical changes of endothelial function in the patients with lymphoproliferative diseases before and after chemotherapy. We performed a search of the clinical results on endothelial function in patients with lymphoproliferative disorders caused by using chemotherapeutic medications from different groups in the PubMed system from 1 January, 2009 to 1 June, 2019 (last ten years), based on the following keywords: endothelium, ED, angiogenesis, endothelin-1 (ET-1), vascular endothelial growth factor (VEGF), cardiovascular events, lymphoproliferative diseases, anthracyclines, alkylating agents. Full-text versions of the articles were evaluated qualitatively and summed up descriptively.
ED and prognostic significance of ET-1 levels
Endothelial cells lining the vessels, including coronary, provide oxygen and free fatty acids vital for the maintenance of metabolic needs of cardiomyocytes, and, as a result, are of great importance for proper functioning of all organs and systems. Mature endothelial cells and their precursors are involved in maintaining physiological homeostasis of cardiac tissue, including regulation of vascular tone and local blood flow, intima permeability and thickness, vascular remodeling and angiogenesis, as well as coagulation and fibrinolysis [12]. Endothelium vasoactive role dysfunction in the regulation of tissue perfusion is caused by imbalance, associated with a decrease in the release of relaxing factors, including nitric oxide, and an increase in the release of contracting factors, including ET-1 (Table 1) [13, 14]. Thus, ED may contribute to limiting blood flow and perfusion of organs and systems. In addition, this complication is of great clinical importance, since it is an initial reversible stage in the development of atherosclerosis [15]. It is proved that ED is the result of endothelium damage, leading to inflammation, activation of endothelial cells, transcription and synthesis of de novo proteins, including adhesion molecules, cytokines, chemokines and procoagulants [16]. Cytokines of inflammation, produced by endothelial cells, are closely related to the development of cardiovascular diseases and, in turn, affect the function of endothelial cells [17, 18]. Damage to endothelial cells can be caused by various factors, such as bacterial or viral infection, impaired regulation of reactive oxygen species, antibodies to endothelial cells, hypoxia, and changes in normal blood circulation or environmental stimuli, including chemotherapy.
Table 1. Endothelium-derived relaxing and contracting factors
|
Vasoconstrictors |
Vasodilators |
|
|
HETE, 20-hydroxyeicosatetraenoic acid; VEGF A, vascular endothelial growth factor A.
The initial response to endothelial damage includes cell signaling and changes in calcium metabolism due to changes in concentrations of nitrogen oxide, prostaglandins, ET-1 and other factors. Then, changes in the basal membrane of smooth muscle cells around endothelial cells appear. Ultimately, vascular remodeling occurs, leading to significant distorders in cell and tissue functioning [19]. According to the proposed mechanism of endothelial damage by cytotoxic agents, vascular changes can be classified into the following groups: direct endothelial damage, activation of coagulation factors, vegetative dysfunction, vasculitis, and fibroblast stimulation. These types of injuries may cause various consequences, including Raynaud's phenomenon, ischemia, heart attack, cerebrovascular attacks, venous thrombosis, thromboembolic complications, and hypertension [1].
The study by Armenian et al., using multivariate regression analysis, discovered a significant increase in the eight-year incidence of cardiovascular diseases among the patients with multiple myeloma, and non-Hodgkin lymphomas compared with an overall population. There was also a significantly increased risk of cardiomyopathy, heart failure and coronary heart disease among the patients treated for CLL, multiple myeloma and non-Hodgkin lymphomas [20]. Skrypnyk et al. described systemic ED in the onset of acute leukemia, which created conditions for occurrence of cardiotoxic effects induced by chemotherapy agents (Figure 1) [21]. It is proved that toxic effect of chemotherapy medications as conditioning agents before autologous bone marrow transplantation in the patients with lymphoproliferative disorders may injure endothelial cells. In particular, fludarabine, often used in conditioning schemes, enhances endothelial cell lysis mediated by cytotoxic lymphocytes [22].

Figure 1. Mechanism of endothelial dysfunction
The toxic effect of anthracyclines on vascular endothelium, implemented via increased oxidative stress, has been recorded. What is important to point out, some authors [23] believe that anthracyclines have a more marked toxicity towards endothelial cells than to blast cells. Anthracycline-induced endothelial toxicity is a complex process influenced by different mechanisms, including pharmaceutical drug accumulation in the nuclei and mitochondria of endotheliocytes (probably due to high affinity for cardiolipin, a negatively charged phospholipid located in the inner membrane of mitochondria) [24, 25], deoxyribonucleic acid repair, stress-induced signaling mechanisms [26, 27], and inhibition of topoisomerase type I and II [25]. In particular, doxorubicin induces the production of hydrogen peroxide, potentiating development of both cardiotoxicity and endothelial toxicity [21]. Considering the fact that in patients with lymphomas, anthracyclines for decades have been the cornerstone of chemotherapy, and their integration into polychemotherapy significantly improved the prognosis of the patients with more aggressive course of the ailment, it is extremely important to diagnose the toxic effects of this group of pharmaceutical drugs [28].
Endothelial damage has also been recorded in the course of administering other chemotherapeutic agents used for treatment of lymphoproliferative disorders, including cyclophosphamide (alkylating agent), and periwinkle alkaloids (depolarizing agents causing distortion of cell microtubules) [12]. Cyclophosphamide generates direct endothelial damage of blood vessels with subsequent transudation of toxic metabolites and damage of myocytes, interstitial hemorrhage, edema and intercapillary microthrombosis. Endothelial toxicity caused by cyclophosphamide, may be due to the pharmacokinetics and metabolism of this medication [29]. These data suggest that various chemotherapy medications cause clinically significant damage of endothelium with a predominant effect directly on endothelial cells. Currently, numerous ED biomarkers are proposed: von Willebrand factor, soluble thrombomodulin, C-reactive protein, cytokines, VEGF, intercellular molecule adhesion-1, ET-1, selectin R and E [30]. However, most of them are not specific to the damage of endothelial cells, and consequently they can resist the function of several types of activated cells, including neutrophils, platelets, mast cells, or macrophages [16], and are not widely used in clinical practice. One of the most specific ED markers is ET-1.
Endothelins, discovered in 1988, are biologically active peptides. Currently, ET-1 and its two isoforms (endothelin-2 and endothelin-3), characterized by a finite set of amino acids, are recognized. ET-1 is formed by proteolytic treatment of preproendothelin (Big-ET), using endothelin-converting enzyme [31]. ET-1 has a very short half-life time. It is formed in the endothelium under the influence of adrenaline, angiotensin-II, vasopressin, thrombin and mechanical impact. In physiological concentrations, it acts on endothelial receptors, stimulating the release of relaxing factors, whereas in higher concentrations, it has a vasospastic effect, causes the proliferation of the media and smooth muscle cells in blood vessels, and potentiates the vasoconstrictor effect of renin-angiotensin-aldosterone system, which aggravates endothelial dysfunction. There is no full understanding of the balance between the vasodilator and vasoconstrictor signals, produced by endothelial cells. Therefore, the question of how this balance changes in blood vessels of different categories and sizes is largely open.
In addition to the role of ET-1 in the regulation of a vascular tone, being 100 times more powerful vasoconstrictor agent than noradrenaline, it is currently in the spotlight as a marker of growth and progression of some tumors. Specifically, preclinical and clinical studies show that ET-1 is involved in tumor cell proliferation, invasion, angiogenesis and neovascularization in different types of tumors [32, 33]. A number of studies have shown that ET-1 and Big-endothelin have prognostic value in cardiac dysfunction and heart attack [34]. Besides, endothelin is a marker of coronary atherosclerosis, coronary ED, liver dysfunction, decreased kidney function. At the same time, some studies noted an increase in ET-1 content at the early stages of lymphoproliferative diseases. For example, a group of researchers, led by Budanova, analyzed the content of ET-1 in 26 patients with lymphoproliferative disorders before and after the completion of the treatment program. The results confirmed that the entire group of patients before vs. after completion of chemotherapy demonstrated strongly increased vs. slightly increased above the norm ET-1 content, correspondingly [35].
Understanding the mechanisms of endotheliocytes chemotherapy is currently a priority problem in connection with lack of a clear algorithm for diagnosis of this complication. Hence, ET-1 content is a promising marker for this ailment. According to Xu et al., elevated concentrations of ET-1 in plasma of patients reflect damage to endothelial cells [36]. In the study by Giordano et al., in patients with acute lymphoblastic leukemia, endothelial damage was noted against the background of chemotherapy. In the follow-up study, a hypothesis was developed that chemotherapy-induced endothelial disorder may contribute to subsequent cardiovascular and metabolic dysfunction observed in a sample of patients in the absence of excess weight. A direct correlation was also found between flow-mediated dilation and ET-1 content. The latter acts as a vasoconstrictor and proinflammatory agent and stimulates mitosis, cell proliferation, free radical formation, and platelet activation [37]. Zver et al. in their examination of the patients with multiple myeloma, after using high doses of cyclophosphamide for treatment, revealed an increased concentration of ET-1 after infusion of cytostatics. They connected their finding with a toxic effect of the medication on vascular endothelium, followed by transudation of toxic metabolites of cyclophosphamide. Simultaneously, the researchers do not deny the fact that the serum content of ET-1 may increase as a result of direct damage to endothelium and neurohormonal activation of heart failure, caused by cardiotoxic action of pharmaceutical drugs [29]. Based on these data, ET-1 is considered an important factor in the development of vascular dysfunction and cardiovascular disease; hence its analysis can be a useful biochemical marker of damage at the stage of subclinical manifestations in patients receiving chemotherapy [38].
Relationship between pathological angiogenesis and VEGF level
Normally, angiogenesis in adults is highly active solely during the healing of tissue damage, menstrual cycles in women and exercise in myocardium and skeletal muscles [39]. The process of angiogenesis is quite complex, and is associated with the interaction of many molecular and cellular elements that control the stability of microenvironment. Inherent elements of the bone marrow microenvironment include hematopoietic, endothelial, immune (e.g., T-lymphocytes), dendritic cells, erythrocytes, osteoblasts and osteoclasts associated with the extracellular matrix. Angiogenic balance in microenvironment depends on effective molecular bonding, mediated by angiogenic factors, such as growth factors, angiogenesis inhibitors, chemokines, cytokines, and other proteins that work in combination to achieve stability.
The most characteristic feature of pathological angiogenesis is stable endothelial cell proliferation. Also, malignant hematopoietic cells secrete angiogenic factors excessively into the microenvironment, thereby stimulating cell growth and multiplication [40]. That is why angiogenesis is a process with a major role in the progression and metastasis of tumors, in particular of oncohematological origin (Figure 2) [41, 42].
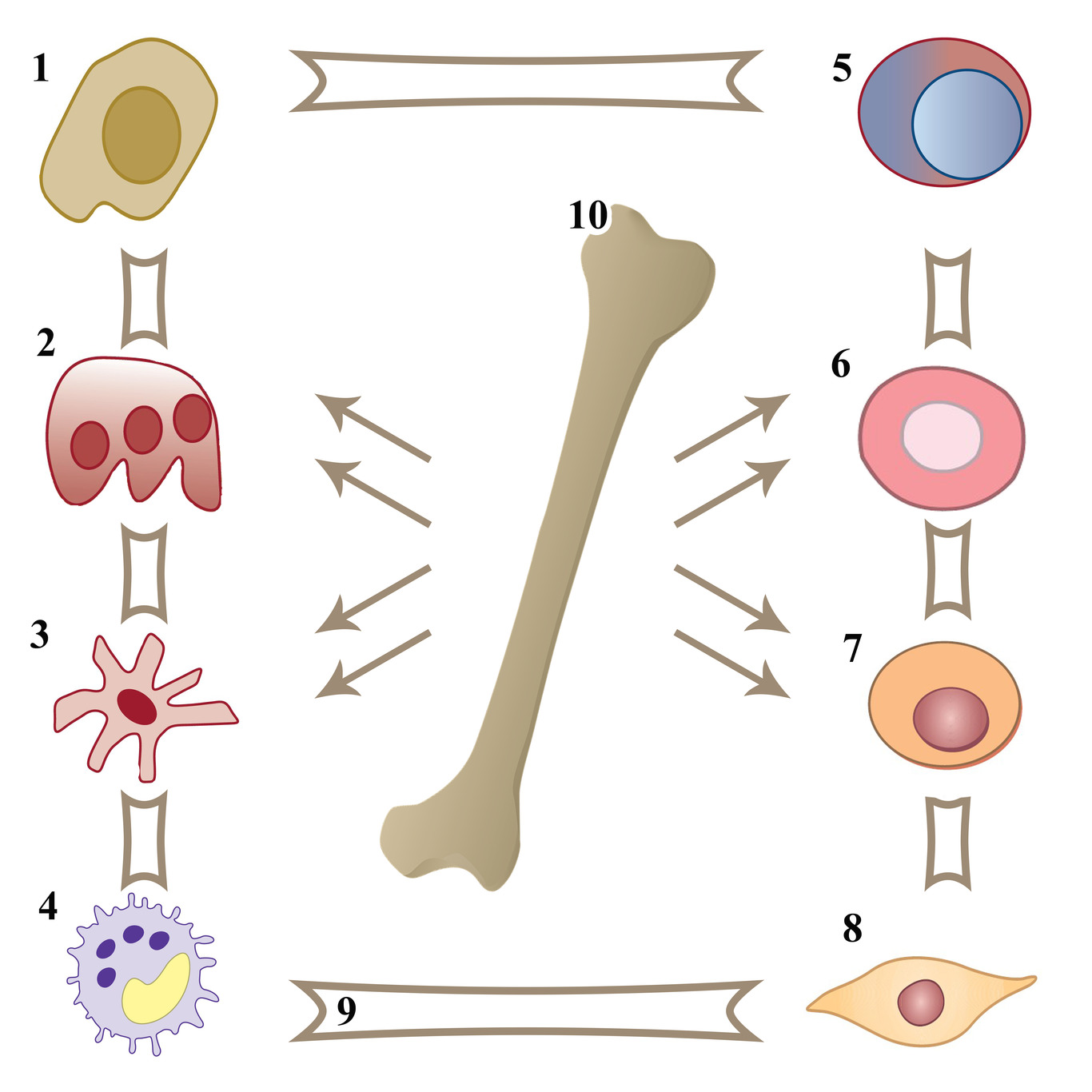
Figure 2. Elements of bone marrow microenvironment and tumor cell involved in pathological angiogenesis. 1 – osteoblast, 2 – osteoclast, 3 – dendritic cell, 4 – tumor cell, 5 – immune cell, 6 – erythrocyte, 7 – hematopoietic cell, 8 – endothelial cell, 9 – extracellular matrix, 10 – bone marrow.
Nowadays, it is widely recognized that angiogenesis promotes the proliferation of malignant cells in CLL [43]; however, the molecular mechanisms of this process are still poorly understood. Tumor substrate cells in CLL produce a large number of angiogenic factors, thus stimulating angiogenesis [44]. Excessive expression of these factors leads to microenvironmental dysfunction, supports proliferation of malignant B-lymphocytes and increases their resistance to apoptosis. Kini et al. in their study reflected the link between pathological angiogenesis and tumor clone in CLL, which revealed a significant rise in the density of microvessels in bone marrow biopsies of the patients, suffering from CLL, compared with control sections [45]. In addition, interleukins 10 and 6, produced by B-lymphocytes in excess of the norm, participate in the angiogenesis stimulation in CLL [46, 47]. Meta-analysis demonstrated that interleukin-10 expression predicted the worst progression-free survival in patients with hematological malignancies [48]. Taking into account that angiogenesis is a complex multicomponent process, it is very important to study it in order to develop treatment strategies and evaluate their effectiveness among patients with lymphoproliferative diseases. One of the most widely studied angiogenic factors in the context of lymphoproliferative diseases is VEGF, so we will focus on this marker.
Because of its biological properties, VEGF is the main mediator of tumor angiogenesis [49]. VEGF is released from endothelial cells located in bone marrow and hematopoietic progenitor cells, and is mostly circulated in platelets and neutrophils. Paracrine VEGF, released by tumor, myeloid, or other stromal cells, enhances the spread of tumor vessels and makes them abnormal, while autocrine VEGF, released by endothelial cells, supports vascular homeostasis [50]. Its presence in threshold concentrations is necessary for endothelial cell survival [51]. A family of structurally related VEGF molecules includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PIGF), main functions of which are presented in Table 2 [52]. The core mediator of tumor angiogenesis is VEGF-A, commonly called VEGF. This indicator stimulates the growth of new blood vessels, providing oxygen and nutrients to the tumor cells, which, in turn, increases tumor proliferative capacity [53]. It is recognized that bone marrow microvascular density is a marker of angiogenesis activity. It exhibits the relationship with the expression, or VEGF serum level, for hematological malignancies [54]. VEGF inherent function is angiogenesis regulation, including angiogenesis of pathological origin, which has been demonstrated in a number of studies [55]. Its expression is associated with proliferation of tumor cells, their invasion and metastasis. Previous studies have established that VEGF is a key cytokine, causing tumor metastasis to local lymph nodes [56]. VEGF may also play an important role in pathogenesis of leukemia, since deregulation of VEGF expression is observed in most patients with oncohematological diseases [3]. Additionally, VEGF is involved in blocking endothelial cell apoptosis, enhancing vascular wall permeability, and developing vasodilation. Some publications describe that natural killer (NK) cells produce various proangiogenic factors in humans and mice [57], such as VEGF and PIGF, which can significantly accelerate tumor growth [58]. In patients suffering from CLL, it was shown that tumor clone cells produce angiogenic factors, including VEGF, stimulating cell proliferation [43, 44].
Table 2. Representatives of the VEGF family and their major functions
|
VEGF subtypes |
Major functions |
|
VEGF-A |
· controls the differentiation and blocking apoptosis of endothelial cells · vasodilation and enhances vascular permeability · regulation of normal and pathological angiogenesis |
|
VEGF-В, PIGF |
· regulation angiogenesis · influences the heart muscle function |
|
VEGF-С, VEGF-D |
· controls lymphangiogenesis · regulates normal angiogenesis at the initial stage and pathological angiogenesis |
VEGF, vascular endothelial growth factor; PIGF, placental growth factor.
A team of scientists led by Maffei examined the patients with CLL and revealed an increase of microvascular density in their bone marrow and affected lymphatic regions. They linked this phenomenon to VEGF-dependent angiogenesis [59]. In addition to the above functions, VEGF regulates the mobility of pathological B-lymphocytes [60] and an interaction among the microenvironment and tumor cells [61]. Andersen et al. demonstrated that CLL patients with higher VEGF content had three times greater risk of ailment progression than those with lower levels of this marker [46]. Dincaslan et al., when examining the patients with non-Hodgkin lymphomas and Hodgkin lymphomas, established that significantly higher VEGF expression was observed in the patients with these nosologies at the onset of the disease, compared with the values recorded when remission was achieved. They associated this finding with decreased tumor impact and lymphangiogenesis activity [53]. Proaggregatory and proinflammatory endothelial stimulation in children with acute lymphoblastic leukemia, according to A. Doroszko et al., is common, while augmented VEGF secretion is aг unreliable indicator of prognosis in short-term follow-up of the children receiving chemotherapy [3].
Despite conflicting results on using VEGF content for diagnostic of chemotherapy toxic effects and pathological angiogenesis, this parameter is of great interest to researchers worldwide, since it has become a promising tool of the targeted therapy of many tumors. However, use of angiogenesis inhibitors, is known to cause various adverse effects on cardiovascular system (Table 3) [62-64], which should be taken into account when making prescription choices.
Table 3. Cardiovascular complications of therapy with angiogenesis inhibitors
|
Medication |
Cardiovascular side effects |
|
Sunitinib [62] |
Heart failure, hypertension |
|
Sorafenib [63] |
Hypertension, ischemia |
|
Bevacizumab [64] |
Heart failure, hypertension, ischemia, thromboembolic arterial complications |
Conclusion
Early detection and timely correction of cardiovascular toxicity are currently the most important tasks in the patients with lymphoproliferative disorders, receiving polychemotherapy. Biomarkers, including ET-1 and VEGF, play a predictive role in diagnosing such changes at early stages. Given the importance of this multidisciplinary problem, it is necessary to continue the search of the most specific biomarkers and methods for timely diagnosis of ED. Thus, understanding the interrelationship among the biological processes under study, the markers and methods used to diagnose them, and the clinical outcomes is essential for improving the quality and duration of life in patients.
Conflict of interests
Authors declare no conflict of interests.
- Soultati A, Mountzios G, Avgerinou C, Papaxoinis G, Pectasides D, Dimopoulos MA, et al. Endothelial vascular toxicity from chemotherapeutic agents: preclinical evidence and clinical implications. Cancer Treat Rev 2012; 38(5): 473-483. https://doi.org/10.1016/j.ctrv.2011.09.002.
- Mikacenic C, Hahn WO, Price BL, Harju-Baker S, Katz R, Kain KC, et al. Biomarkers of Endothelial Activation Are Associated with Poor Outcome in Critical Illness. PLoS One 2015; 10(10): e0141251. https://doi.org/10.1371/journal.pone.0141251.
- Doroszko A, Niedzielska E, Jakubowski M, Porwolik J, Turek-Jakubowska A, Szahidewicz-Krupska E, et al. Endothelial Function in Children with Acute Lymphoblastic Leukemia (ALL) May Reflect the Clinical Outcome. Biomed Res Int 2018; 2018: 7918091. https://doi.org/10.1155/2018/7918091.
- Cardinale D, Bacchiani G, Beggiato M, Colombo A, Cipolla CM. Strategies to prevent and treat cardiovascular risk in cancer patients. Semin Oncol 2013; 40(2): 186-198. https://doi.org/10.1053/j.seminoncol.2013.01.008.
- Lipshultz SE, Adams MJ, Colan SD, Constine LS, Herman EH, Hsu DT, et al; American Heart Association Congenital Heart Defects Committee of the Council on Cardiovascular Disease in the Young, Council on Basic Cardiovascular Sciences, Council on Cardiovascular and Stroke Nursing, Council on Cardiovascular Radiolo. Long-term cardiovascular toxicity in children, adolescents, and young adults who receive cancer therapy: pathophysiology, course, monitoring, management, prevention, and research directions: a scientific statement from the American Heart Association. Circulation 2013; 128(17): 1927-1995. https://doi.org/10.1161/cir.0b013e3182a88099.
- Tabrizi R, Vakili S, Lankarani KB, Akbari M, Jamilian M, Mahdizadeh Z, et al. The Effects of Vitamin D Supplementation on Markers Related to Endothelial Function Among Patients with Metabolic Syndrome and Related Disorders: A Systematic Review and Meta-Analysis of Clinical Trials. Horm Metab Res 2018; 50(8): 587-596. https://doi.org/10.1055/a-0651-4842.
- Shahini L, Gašparov S, Petruševska G, Manxhuka Kerliu S, Veselaj F, Kurshumliu F, et al. Clinical Significance of VEGF-A and Microvessel Density in Diffuse Large B-Cell Lymphoma and Low-Grade Follicular Lymphoma. Acta Clin Croat 2017; 56(4): 588-593. https://doi.org/10.20471/acc.2017.56.04.02.
- Fowler N, Davis E. Targeting B-cell receptor signaling: changing the paradigm. Hematology Am Soc Hematol Educ Program 2013; 2013: 553-560. https://doi.org/10.1182/asheducation-2013.1.553.
- Fruchon S, Kheirallah S, Al Saati T, Ysebaert L, Laurent C, Leseux L, et al. Involvement of the Syk-mTOR pathway in follicular lymphoma cell invasion and angiogenesis. Leukemia 2012; 26(4): 795-805. https://doi.org/10.1038/leu.2011.248.
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64(1): 9-29. https://doi.org/10.3322/caac.21208.
- Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013; 369(1): 32-42. https://doi.org/10.1056/nejmoa1215637.
- Morbidelli L, Donnini S, Ziche M. Targeting endothelial cell metabolism for cardio-protection from the toxicity of antitumor agents. Cardio-Oncol 2016; 2: 1-12. https://doi.org/10.1186/s40959-016-0010-6.
- Zhang J, Defelice AF, Hanig JP, Colatsky T. Biomarkers of endothelial cell activation serve as potential surrogate markers for drug-induced vascular injury. Toxicol Pathol 2010; 38(6): 856-871. https://doi.org/10.1177/0192623310378866.
- Su JB. Vascular endothelial dysfunction and pharmacological treatment. World J Cardiol 2015; 7(11): 719-741. https://doi.org/10.4330/wjc.v7.i11.719.
- Beaudry RI, Liang Y, Boyton ST, Tucker WJ, Brothers RM, Daniel KM, et al. Meta-analysis of Exercise Training on Vascular Endothelial Function in Cancer Survivors. Integr Cancer Ther 2018; 17(2): 192-199. https://doi.org/10.1177/1534735418756193.
- D'Andrea S, Barbonetti A, Martorella A, Necozione S, Francavilla F, Francavilla S. Effect of prolonged treatment with phosphodiesterase-5-inhibitors on endothelial dysfunction in vascular diseases and vascular risk conditions: A systematic review analysis and meta-analysis of randomized double-blind placebo-controlled trials. Int J Clin Pract 2019; 73(2): e13296. https://doi.org/10.1111/ijcp.13296.
- Zhao T, Kecheng Y, Zhao X, Hu X, Zhu J, Wang Y, et al. The higher serum endocan levels may be a risk factor for the onset of cardiovascular disease: A meta-analysis. Medicine (Baltimore) 2018; 97(49): e13407. https://doi.org/10.1097/md.0000000000013407.
- Schwingshackl L, Christoph M, Hoffmann G. Effects of Olive Oil on Markers of Inflammation and Endothelial Function-A Systematic Review and Meta-Analysis. Nutrients 2015; 7(9): 7651-7675. https://doi.org/10.3390/nu7095356.
- Vion AC, Rautou PE, Durand F, Boulanger CM, Valla DC. Interplay of Inflammation and Endothelial Dysfunction in Bone Marrow Transplantation: Focus on Hepatic Veno-Occlusive Disease. Semin Thromb Hemost 2015; 41(6): 629-643. https://doi.org/10.1055/s-0035-1556728.
- Armenian SH, Xu L, Ky B, Sun C, Farol LT, Pal SK, et al. Cardiovascular Disease Among Survivors of Adult-Onset Cancer: A Community-Based Retrospective Cohort Study. J Clin Oncol 2016; 34(10): 1122-1130. https://doi.org/10.1200/jco.2015.64.0409.
- Skrypnyk I, Maslova G, Lymanets T, Gusachenko I. L-arginine is an effective medication for prevention of endothelial dysfunction, a predictor of anthracycline cardiotoxicity in patients with acute leukemia. Exp Oncol 2017; 39(4): 308-311. https://pubmed.ncbi.nlm.nih.gov/29284775.
- Blix ES, Husebekk A. Raiders of the lost mark – endothelial cells and their role in transplantation for hematologic malignancies. Leuk Lymphoma 2016; 57(12): 2752-2762. https://doi.org/10.1080/10428194.2016.1201566.
- Ajithkumar GS, Ramachandran S, Kartha CC. Drug induced endothelial dysfunction: functional role of oxidative stress. IIOABJ 2011; 2(5): 62-70. http://rgcb.sciencecentral.in/583.
- Majzner K, Wo´jcik T, Szafraniec E, Łukawska M, Oszczapowicz I, Chlopicki S, et al. Nuclear accumulation of anthracyclines in the endothelium studied by bimodal imaging: fluorescence and Raman microscopy. Analyst 2015; 140(7): 2302-2310. https://doi.org/10.1039/c4an01882f.
- Volkova M, Russell R 3rd. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev 2011; 7(4): 214-220. https://doi.org/10.2174/157340311799960645.
- Feng R, Zhai WL, Yang HY, Jin H, Zhang QX. Induction of ER stress protects gastric cancer cells against apoptosis induced by cisplatin and doxorubicin through activation of p38 MAPK. Biochem Biophys Res Commun 2011; 406(2): 299-304. https://doi.org/10.1016/j.bbrc.2011.02.036.
- Diers AR, Broniowska KA, Hogg N. Nitrosative stress and redox-cycling agents synergize to cause mitochondrial dysfunction and cell death in endothelial cells. Redox Biol 2013; 1(1): 1-7. https://doi.org/10.1016/j.redox.2012.11.003.
- Mele D, Nardozza M, Spallarossa P, Frassoldati A, Tocchetti CG, Cadeddu C, et al. Current views on anthracycline cardiotoxicity. Heart Fail Rev 2016; 21(5): 621-634. https://doi.org/10.1007/s10741-016-9564-5.
- Zver S, Zadnik V, Černelč P, Koželj M. Cardiac toxicity of high-dose cyclophosphamide and melphalan in patients with multiple myeloma treated with tandem autologous hematopoietic stem cell transplantation. Int J Hematol 2008; 88(2): 227-236. https://doi.org/10.1007/s12185-008-0112-5.
- Smiljic S. The clinical significance of endocardial endothelial dysfunction. Medicina (Kaunas) 2017; 53(5): 295-302. https://doi.org/10.1016/j.medici.2017.08.003.
- Zhang H, Li Y, Zeng Y, Wu R, Ou J. Endothelin-1 downregulates angiotensin-converting enzyme-2 expression in human bronchial epithelial cells. Pharmacology 2013; 91(5-6): 297-304. https://doi.org/10.1159/000350395.
- Kalles V, Zografos GC, Provatopoulou X, Kalogera E, Liakou P, Georgiou G, et al. Circulating levels of endothelin-1 (ET-1) and its precursor (Big ET-1) in breast cancer early diagnosis. Tumour Biol 2012; 33(4): 1231-1236. https://doi.org/10.1007/s13277-012-0371-x.
- Stow LR, Jacobs ME, Wingo CS, Cain BD. Endothelin-1 gene regulation. FASEB J 2011; 25(1): 16-28. https://doi.org/10.1096/fj.10-161612.
- Wennberg P, Wensley F, Di Angelantonio E, Johansson L, Boman K, Rumley A, et al. Haemostatic and inflammatory markers are independently associated with myocardial infarction in men and women. Thromb Res 2012; 129(1): 68-73. https://doi.org/10.1016/j.thromres.2011.05.015.
- Budanova DA, Belenkov YN, Sokolova IY, Antyufeeva ON, Ershov VI, Ilgisonis IS, et al. The Role of Endothelial Dysfunction in the Development of Cardiotoxic Action of Cytostatics in Patients with Lymphoproliferative Diseases. Kardiologiia 2019; 59(4): 64-66. Russian. https://doi.org/10.18087/cardio.2019.4.10251.
- Xu M, Lu YP, Hasan AA, Hocher B. Plasma ET-1 Concentrations are Elevated in Patients with Hypertension – Meta-Analysis of Clinical Studies. Kidney Blood Press Res 2017; 42(2): 304-313. https://doi.org/10.1159/000477572.
- Giordano P, Molinari AC, Del Vecchio GC, Saracco P, Russo G, Altomare M, et al. Prospective study of hemostatic alterations in children with acute lymphoblastic leukemia. Am J Hematol 2010; 85(5): 325-330. https://doi.org/10.1002/ajh.21665.
- Giordano P, Muggeo P, Delvecchio M, Carbonara S, Romano A, Altomare M, et al. Endothelial dysfunction and cardiovascular risk factors in childhood acute lymphoblastic leukemia survivors. Int J Cardiol 2017; 228: 621-627. https://doi.org/10.1016/j.ijcard.2016.11.025.
- De Falco S. The discovery of placenta growth factor and its biological activity. Exp Mol Med 2012; 44(1): 1-9. https://doi.org/10.3858/emm.2012.44.1.025.
- Aguirre Palma LM, Gehrke I, Kreuzer KA. Angiogenic factors in chronic lymphocytic leukaemia (CLL): Where do we stand? Crit Rev Oncol Hematol 2015; 93(3): 225-236. https://doi.org/10.1016/j.critrevonc.2014.10.007.
- Aggarwal D, Srivastava G, Gupta R, Pant L, Krishan G, Singh S. Angiogenesis in non-Hodgkin lymphoma: an intercategory comparison of microvessel density. ISRN Hematol 2012; 2012: 943089. https://doi.org/10.5402/2012/943089.
- Negaard HF, Iversen N, Bowitz-Lothe IM, Sandset PM, Steinsvik B, Ostenstad B, et al. Increased bone marrow microvascular density in haematological malignancies is associated with differential regulation of angiogenic factors. Leukemia 2009; 23(1): 162-169. https://doi.org/10.1038/leu.2008.255.
- Piechnik A, Dmoszynska A, Omiotek M, Mlak R, Kowal M, Stilgenbauer S, et al. The VEGF receptor, neuropilin-1, represents a promising novel target for chronic lymphocytic leukemia patients. Int J Cancer 2013; 133(6): 1489-1496. https://doi.org/10.1002/ijc.28135.
- Aguirre Palma LM, Flamme H, Gerke I, Kreuzer KA. Angiopoietins modulate survival, migration, and the components of the Ang-Tie2 pathway of chronic lymphocytic leukaemia (CLL) cells in vitro. Cancer Microenviron 2016; 9(1): 13-26. https://doi.org/10.1007/s12307-016-0180-7.
- Kini AR, Kay NE, Peterson LC. Increased bone marrow angiogenesis in B cell chronic lymphocytic leukemia. Leukemia 2000; 14(8): 1414-1418. https://doi.org/10.1038/sj.leu.2401825.
- Andersen BL, Goyal NG, Weiss DM, Westbrook TD, Maddocks KJ, Byrd JC, et al. Cells, cytokines, chemokines, and cancer stress: A biobehavioral study of patients with chronic lymphocytic leukemia. Cancer 2018; 124(15): 3240-3248. https://doi.org/10.1002/cncr.31538.
- Alhakeem SS, Sindhava VJ, McKenna MK, Gachuki BW, Byrd JC, Muthusamy N, et al. Role of B cell receptor signaling in IL-10 production by normal and malignant B-1 cells. Ann N Y Acad Sci 2015; 1362(1): 239-249. https://doi.org/10.1111/nyas.12802.
- Zhao S, Wu D, Wu P, Wang Z, Huang J. Serum IL-10 Predicts Worse Outcome in Cancer Patients: A Meta-Analysis. PLoS One 2015; 10(10): e0139598. https://doi.org/10.1371/journal.pone.0139598.
- Li J, Gu J. Cardiovascular Toxicities with Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer Patients: A Meta-Analysis of 77 Randomized Controlled Trials. Clin Drug Investig 2018; 38(12): 1109-1123. https://doi.org/10.1007/s40261-018-0709-2.
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011; 473(7347): 298-307. https://doi.org/10.1038/nature10144.
- Fang Y, Wu D, Birukov KG. Mechanosensing and Mechanoregulation of Endothelial Cell Functions. Compr Physiol 2019; 9(2): 873-904. https://doi.org/10.1002/cphy.c180020.
- Saberi-Karimian M, Katsiki N, Caraglia M, Boccellino M, Majeed M, Sahebkar A. Vascular endothelial growth factor: An important molecular target of curcumin. Crit Rev Food Sci Nutr 2019; 59(2): 299-312. https://doi.org/10.1080/10408398.2017.1366892.
- Geng L, Chaudhuri A, Talmon G, Wisecarver JL, Wang J. TGF-beta suppresses VEGFA-mediated angiogenesis in colon cancer metastasis. PLoS One 2013; 8(3): e59918. https://doi.org/10.1371/journal.pone.0059918.
- Dincaslan HU, Yavuz G, Unal E, Tacyildiz N, Ikinciogullari A, Dogu F, et al. Does serum soluble vascular endothelial growth factor levels have different importance in pediatric acute leukemia and malignant lymphoma patients? Pediatr Hematol Oncol 2010; 27(7): 503-516. https://doi.org/10.3109/08880018.2010.493574.
- Ferrara N. VEGF-A: a critical regulator of blood vessel growth. Eur Cytokine Netw 2009; 20(4): 158-163. https://doi.org/10.1684/ecn.2009.0170.
- Su F, Liu B, Chen M, Xiao J, Li X, Lv X, et al. Association between VEGF-A, C and D expression and lymph node involvement in breast cancer: a meta-analysis. Int J Biol Markers 2016; 31(3): e235-244. https://doi.org/10.5301/jbm.5000198.
- Vacca P, Moretta L, Moretta A, Mingari MC. Origin, phenotype and function of human natural killer cells in pregnancy. Trends Immunol 2011; 32(11): 517-523. https://doi.org/10.1016/j.it.2011.06.013.
- Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol 2012; 12(4): 239-252. https://doi.org/10.1038/nri3174.
- Maffei R, Martinelli S, Castelli I, Santachiara R, Zucchini P, Fontana M, et al. Increased angiogenesis induced by chronic lymphocytic leukemia B cells is mediated by leukemia-derived Ang2 and VEGF. Leuk Res 2010; 34(3): 312-321. https://doi.org/10.1016/j.leukres.2009.06.023.
- Ugarte-Berzal E, Redondo-Muñoz J, Eroles P, Del Cerro MH, García-Marco JA, Terol MJ, et al. VEGF/VEGFR2 interaction down-regulates matrix metalloproteinase-9 via STAT1 activation and inhibits B chronic lymphocytic leukemia cell migration. Blood 2010; 115(4): 846-849. https://doi.org/10.1182/blood-2009-08-239426.
- Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood 2010; 115(9): 1755-1764. https://doi.org/10.1182/blood-2009-09-242719.
- Gore ME, Szczylik C, Porta C, Bracarda S, Bjarnason GA, Oudard S, et al. Final results from the large sunitinib global expanded-access trial in metastatic renal cell carcinoma. Br J Cancer 2015; 113(1): 12-19. https://doi.org/10.1038/bjc.2015.196.
- Rini B, Szczylik C, Tannir NM, Koralewski P, Tomczak P, Deptala A, et al. AMG 386 in combination with sorafenib in patients with metastatic clear cell carcinoma of the kidney: a randomized, double-blind, placebo-controlled, phase 2 study. Cancer 118(24): 6152-6161. https://doi.org/10.1002/cncr.27632.
- Beck J, Procopio G, Bajetta E, Keilholz U, Negrier S, Szczylik C, et al. Final results of the European Advanced Renal Cell Carcinoma Sorafenib (EU-ARCCS) expanded-access study: a large open-label study in diverse community settings. Ann Oncol 2011; 22(8): 1812-1823. https://doi.org/10.1093/annonc/mdq651.
Received 9 November 2019, Revised 27 May 2020, Accepted 15 June 2020
© 2019, Davydkin I.L., Kuzmina T.P., Naumova K.V., Khayretdinov R.K., Danilova O.E., Stepanova T.Yu., Osadchuk A.M., Mordvinova E.V.
© 2019, Russian Open Medical Journal
Correspondence to Tatyana P. Kuzmina. Phone: +7(927)749-64-47. E-mail: tatyana_kuzmina_91@bk.ru.